

The genetic basis of aortic aneurysm
- PMID: 25183854
- PMCID: PMC4143103
- DOI: 10.1101/cshperspect.a015909
The genetic basis of aortic aneurysm
Abstract
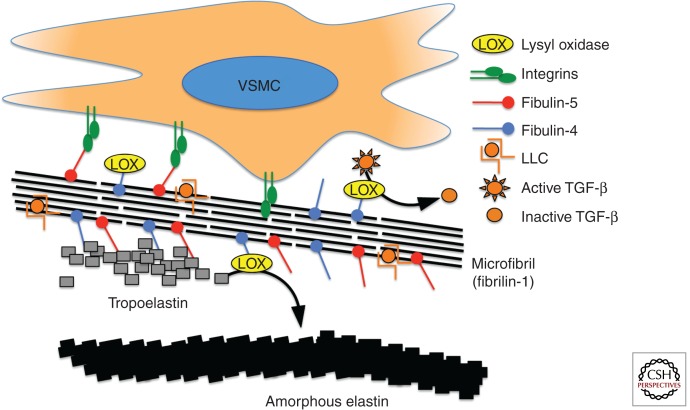
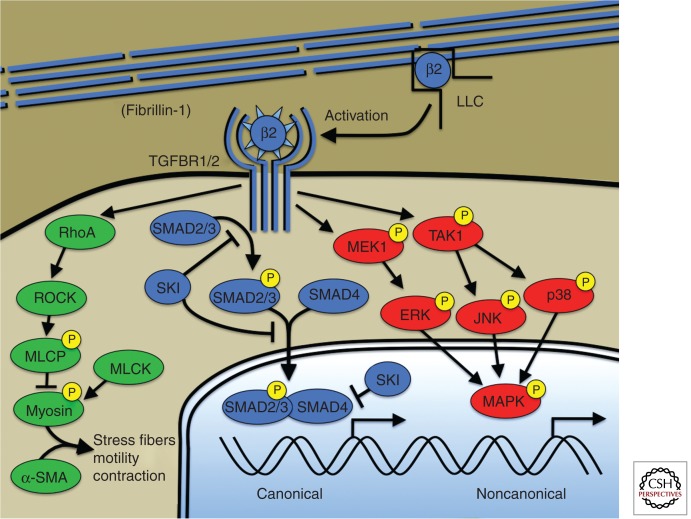
Gene identification in human aortic aneurysm conditions is proceeding at a rapid pace and the integration of pathogenesis-based management strategies in clinical practice is an emerging reality. Human genetic alterations causing aneurysm involve diverse gene products including constituents of the extracellular matrix, cell surface receptors, intracellular signaling molecules, and elements of the contractile cytoskeleton. Animal modeling experiments and human genetic discoveries have extensively implicated the transforming growth factor-β (TGF-β) cytokine-signaling cascade in aneurysm progression, but mechanistic links between many gene products remain obscure. This chapter will integrate human genetic alterations associated with aortic aneurysm with current basic research findings in an attempt to form a reconciling if not unifying model for hereditary aortic aneurysm.
Copyright © 2014 Cold Spring Harbor Laboratory Press; all rights reserved.
Figures
References
-
- Andrabi S, Bekheirnia MR, Robbins-Furman P, Lewis RA, Prior TW, Potocki L 2011. SMAD4 mutation segregating in a family with juvenile polyposis, aortopathy, and mitral valve dysfunction. Am J Med Genet A 155A: 1165–1169 - PubMed
-
- Bergen AA, Plomp AS, Schuurman EJ, Terry S, Breuning M, Dauwerse H, Swart J, Kool M, van Soest S, Baas F, et al. 2000. Mutations in ABCC6 cause pseudoxanthoma elasticum. Nat Genet 25: 228–231 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical