

Tackling the tumor microenvironment: what challenge does it pose to anticancer therapies?
- PMID: 25185441
- PMCID: PMC4225463
- DOI: 10.1007/s13238-014-0097-1
Tackling the tumor microenvironment: what challenge does it pose to anticancer therapies?
Abstract
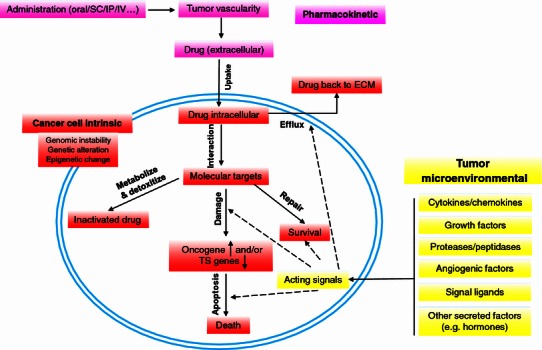
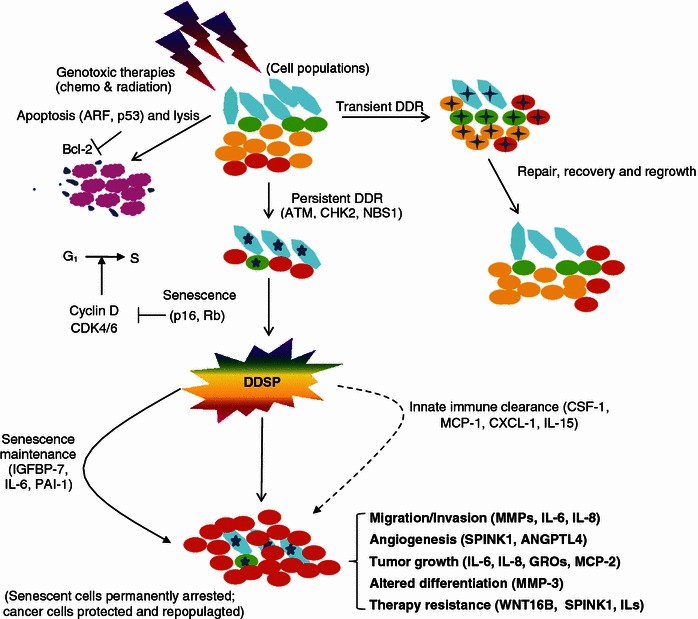
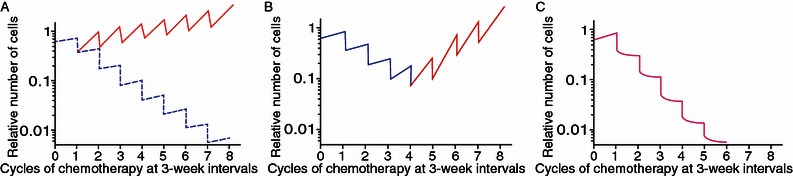
Cancer is a highly aggressive and devastating disease, and impediments to a cure arise not just from cancer itself. Targeted therapies are difficult to achieve since the majority of cancers are more intricate than ever imagined. Mainstream methodologies including chemotherapy and radiotherapy as routine clinical regimens frequently fail, eventually leading to pathologies that are refractory and incurable. One major cause is the gradual to rapid repopulation of surviving cancer cells during intervals of multiple-dose administration. Novel stress-responsive molecular pathways are increasingly unmasked and show promise as emerging targets for advanced strategies that aim at both de novo and acquired resistance. We highlight recent data reporting that treatments particularly those genotoxic can induce highly conserved damage responses in non-cancerous constituents of the tumor microenvironment (TMEN). Master regulators, including but not limited to NF-kB and C/EBP-β, are implicated and their signal cascades culminate in a robust, chronic and genome-wide secretory program, forming an activated TMEN that releases a myriad of soluble factors. The damage-elicited but essentially off target and cell non-autonomous secretory phenotype of host stroma causes adverse consequences, among which is acquired resistance of cancer cells. Harnessing signals arising from the TMEN, a pathophysiological niche frequently damaged by medical interventions, has the potential to promote overall efficacy and improve clinical outcomes provided that appropriate actions are ingeniously integrated into contemporary therapies. Thereby, anticancer regimens should be well tuned to establish an innovative clinical avenue, and such advancement will allow future oncological treatments to be more specific, accurate, thorough and personalized.
Figures
References
-
- Acosta JC, O'Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, Fumagalli M, Da Costa M, Brown C, Popov N, Takatsu Y, Melamed J, d'Adda di Fagagna F, Bernard D, Hernando E, Gil J (2008) Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 133:1006–1018 - PubMed
-
- Alspach E, Flanagan KC, Luo X, Ruhland MK, Huang H, Pazolli E, Donlin MJ, Marsh T, Piwnica-Worms D, Monahan J, Novack DV, McAllister SS, Stewart SA. p38MAPK plays a crucial role in stromal-mediated tumorigenesis. Cancer Discov. 2014;4:716–729. doi: 10.1158/2159-8290.CD-13-0743. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
