

Effective diagnosis of genetic disease by computational phenotype analysis of the disease-associated genome
- PMID: 25186178
- PMCID: PMC4512639
- DOI: 10.1126/scitranslmed.3009262
Effective diagnosis of genetic disease by computational phenotype analysis of the disease-associated genome
Abstract
Less than half of patients with suspected genetic disease receive a molecular diagnosis. We have therefore integrated next-generation sequencing (NGS), bioinformatics, and clinical data into an effective diagnostic workflow. We used variants in the 2741 established Mendelian disease genes [the disease-associated genome (DAG)] to develop a targeted enrichment DAG panel (7.1 Mb), which achieves a coverage of 20-fold or better for 98% of bases. Furthermore, we established a computational method [Phenotypic Interpretation of eXomes (PhenIX)] that evaluated and ranked variants based on pathogenicity and semantic similarity of patients' phenotype described by Human Phenotype Ontology (HPO) terms to those of 3991 Mendelian diseases. In computer simulations, ranking genes based on the variant score put the true gene in first place less than 5% of the time; PhenIX placed the correct gene in first place more than 86% of the time. In a retrospective test of PhenIX on 52 patients with previously identified mutations and known diagnoses, the correct gene achieved a mean rank of 2.1. In a prospective study on 40 individuals without a diagnosis, PhenIX analysis enabled a diagnosis in 11 cases (28%, at a mean rank of 2.4). Thus, the NGS of the DAG followed by phenotype-driven bioinformatic analysis allows quick and effective differential diagnostics in medical genetics.
Copyright © 2014, American Association for the Advancement of Science.
Conflict of interest statement
Competing interests: Y.M. is an equity holder of Cartagenia NV, and S.M. is a paid consultant for Agilent. The other authors declare that they have no competing interests.
Figures
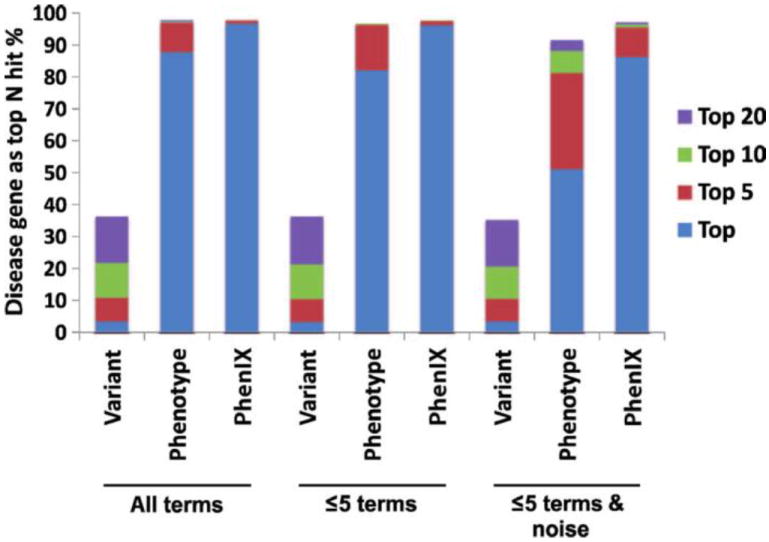
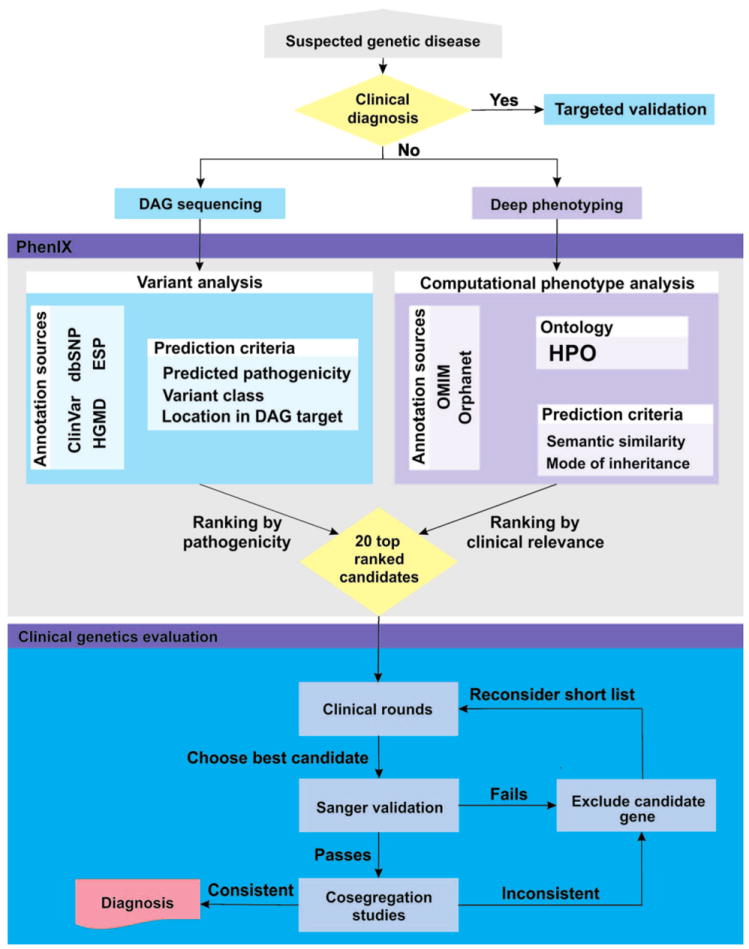
References
-
- Amberger J, Bocchini C, Hamosh A. A new face and new challenges for Online Mendelian Inheritance in Man (OMIM®) Hum Mutat. 2011;32:564–567. - PubMed
-
- Rath A, Olry A, Dhombres F, Brandt MM, Urbero B, Ayme S. Representation of rare diseases in health information systems: The Orphanet approach to serve a wide range of end users. Hum Mutat. 2012;33:803–808. - PubMed
-
- Köhler S, Doelken SC, Mungall CJ, Bauer S, Firth HV, Bailleul-Forestier I, Black GC, Brown DL, Brudno M, Campbell J, FitzPatrick DR, Eppig JT, Jackson AP, Freson K, Girdea M, Helbig I, Hurst JA, Jähn J, Jackson LG, Kelly AM, Ledbetter DH, Mansour S, Martin CL, Moss C, Mumford A, Ouwehand WH, Park SM, Riggs ER, Scott RH, Sisodiya S, Van Vooren S, Wapner RJ, Wilkie AO, Wright CF, Vulto-van Silfhout AT, de Leeuw N, de Vries BB, Washingthon NL, Smith CL, Westerfield M, Schofield P, Ruef BJ, Gkoutos GV, Haendel M, Smedley D, Lewis SE, Robinson PN. The Human Phenotype Ontology project: Linking molecular biology and disease through phenotype data. Nucleic Acids Res. 2014;42:D966–D974. - PMC - PubMed
-
- Shashi V, McConkie-Rosell A, Rosell B, Schoch K, Vellore K, McDonald M, Jiang YH, Xie P, Need A, Goldstein DB. The utility of the traditional medical genetics diagnostic evaluation in the context of next-generation sequencing for undiagnosed genetic disorders. Genet Med. 2014;16:176–182. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
