

Chemical induction of unfolded protein response enhances cancer cell killing through lytic virus infection
- PMID: 25187554
- PMCID: PMC4249087
- DOI: 10.1128/JVI.02156-14
Chemical induction of unfolded protein response enhances cancer cell killing through lytic virus infection
Abstract
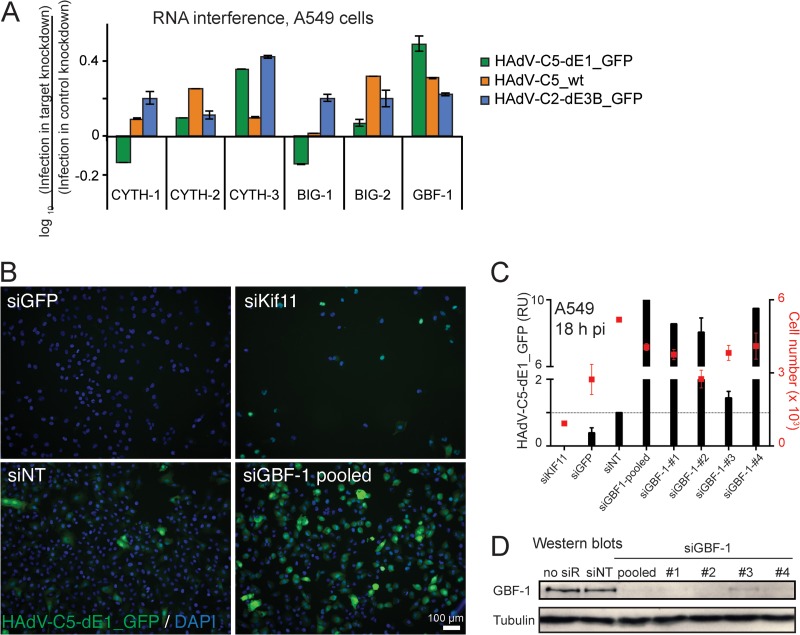
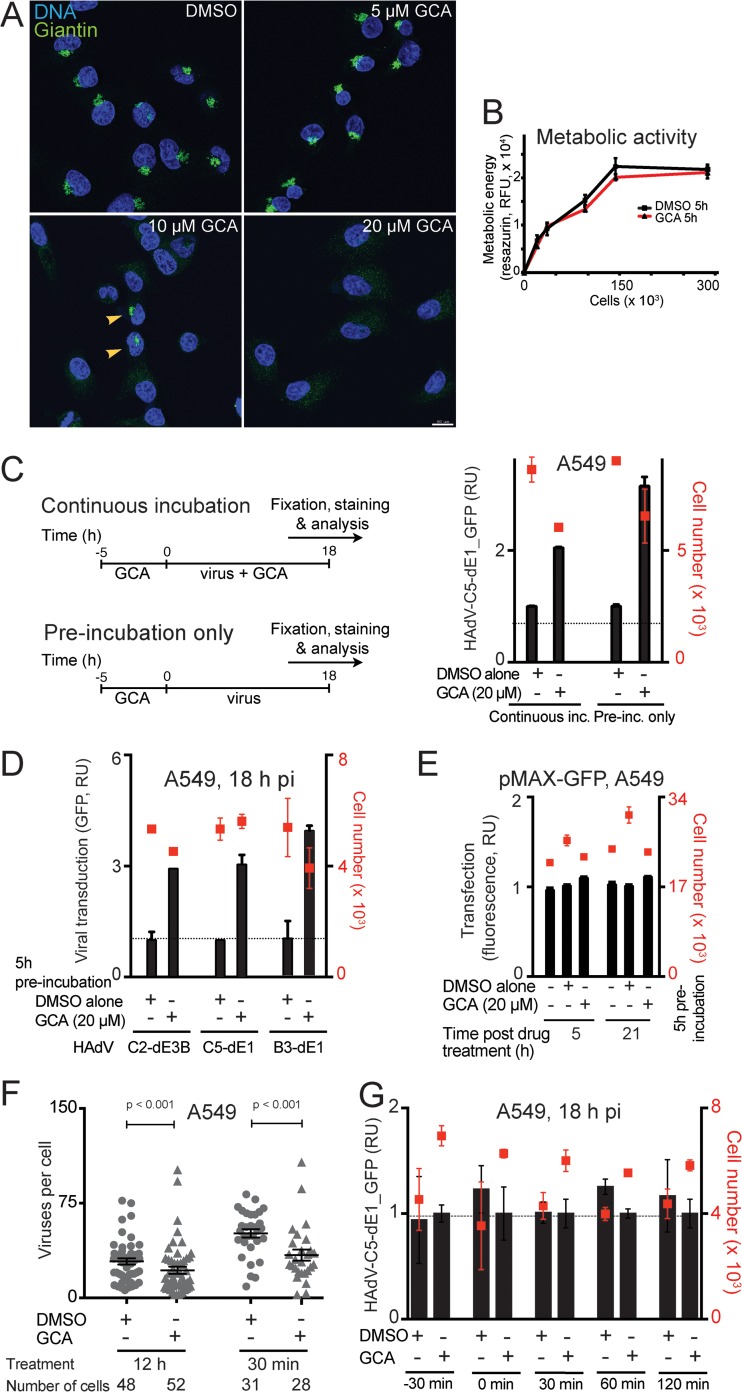
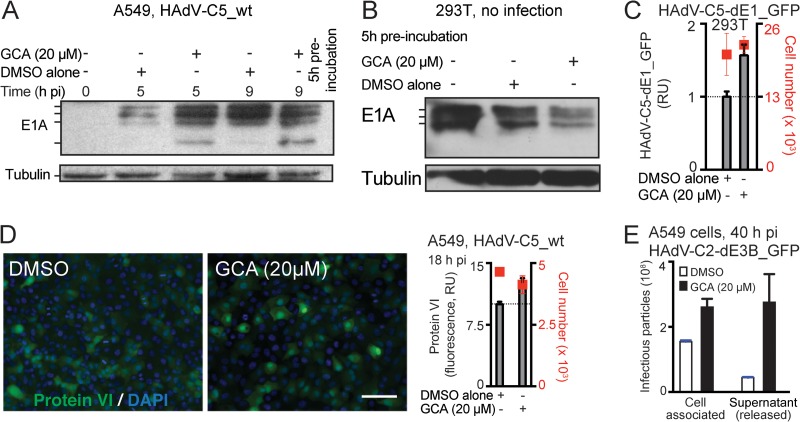
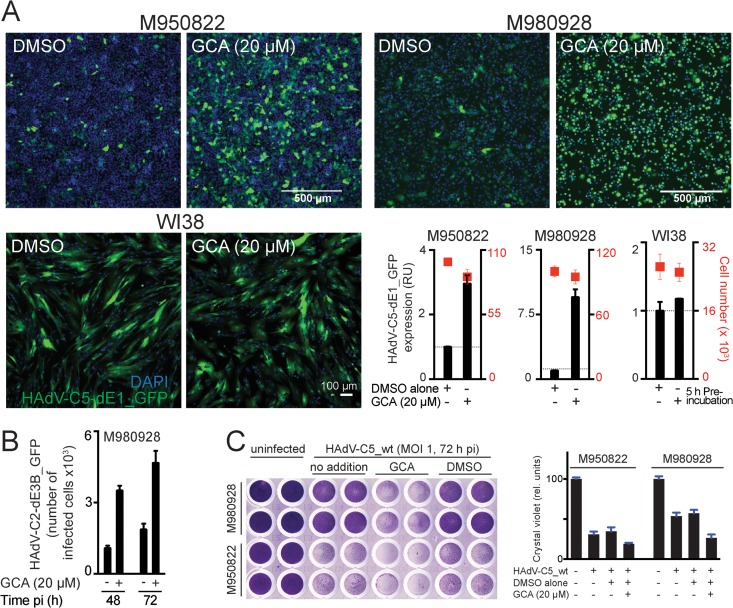
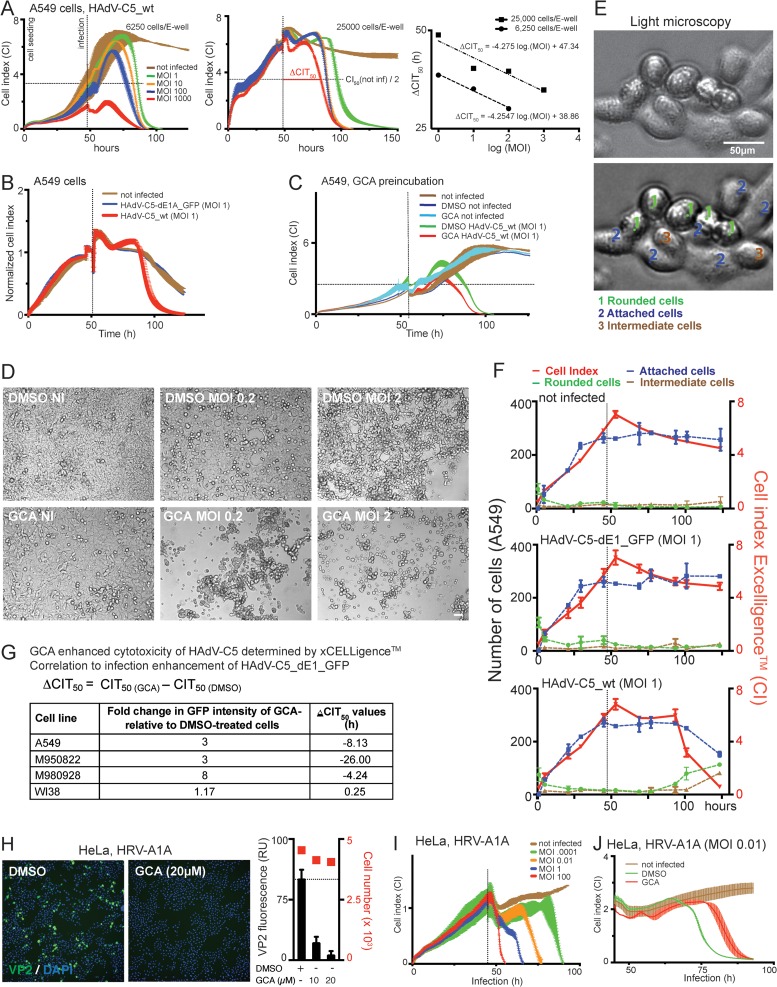
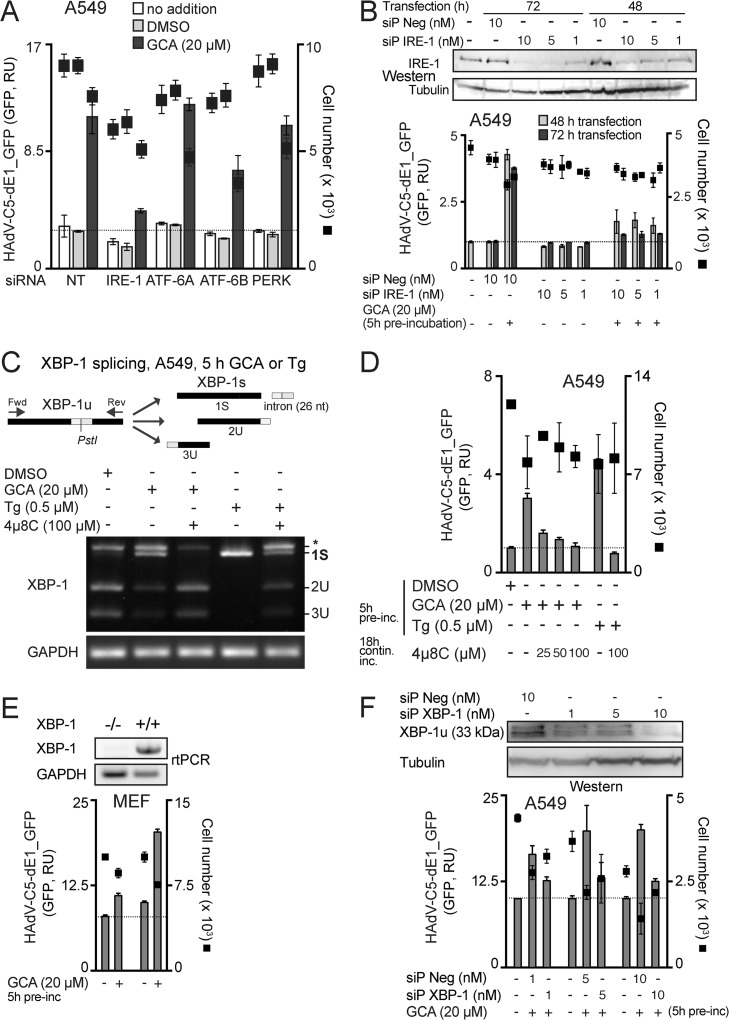
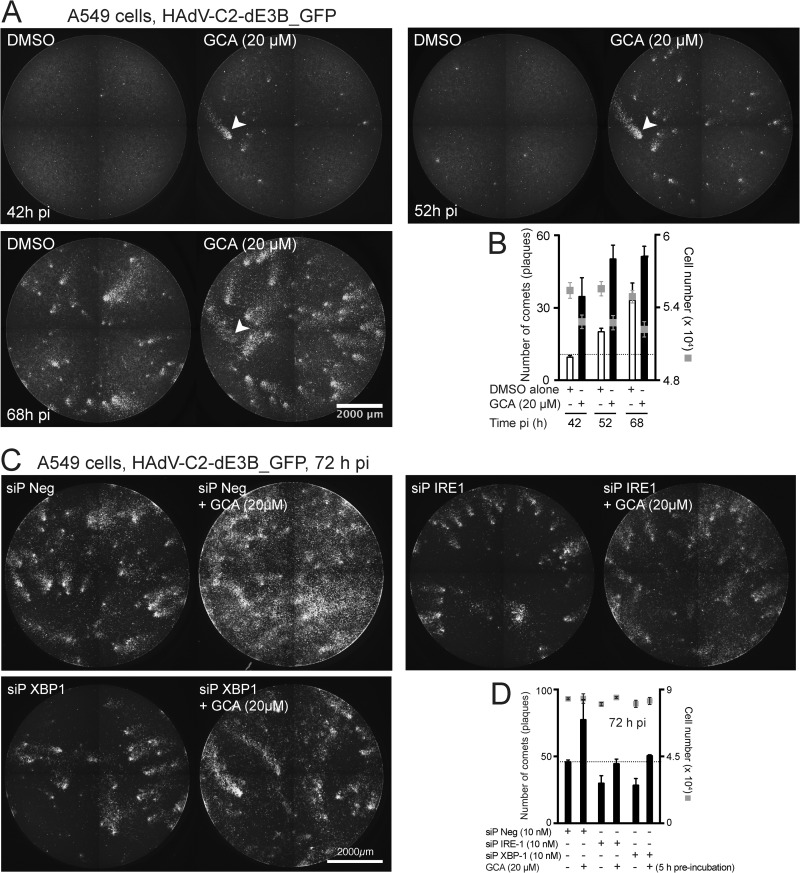
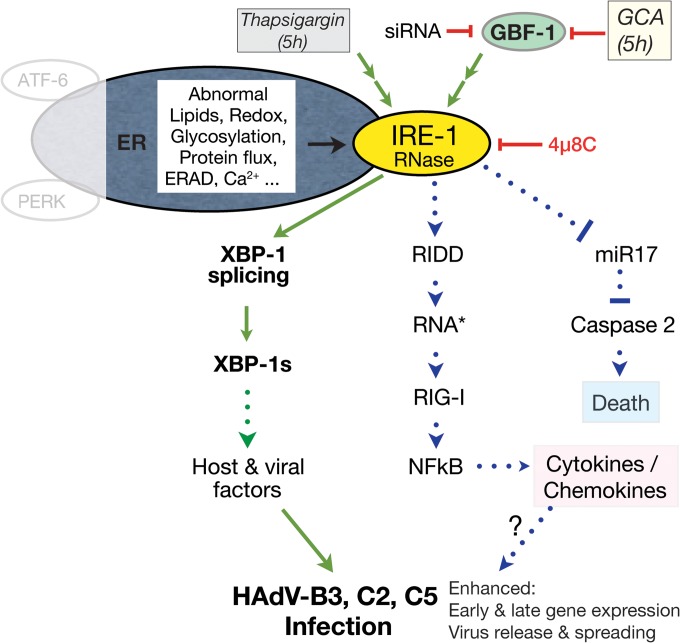
Cancer cells are susceptible to oncolytic viruses, albeit variably. Human adenoviruses (HAdVs) are widely used oncolytic agents that have been engineered to produce progeny within the tumor and elicit bystander effects. We searched for host factors enhancing bystander effects and conducted a targeted RNA interference screen against guanine nucleotide exchange factors (GEFs) of small GTPases. We show that the unfolded protein response (UPR), which is readily inducible in aggressive tumor cells, enhances melanoma or epithelial cancer cell killing upon HAdV infection. UPR was triggered by knockdown of Golgi-specific brefeldin A-resistant guanine nucleotide exchange factor 1 (GBF-1) or the GBF-1 inhibitor golgicide A (GCA) and stimulated HAdV infection. GBF-1 is a GEF for ADP ribosylation factors (Arfs) regulating endoplasmic reticulum (ER)-to-Golgi apparatus and intra-Golgi apparatus membrane transport. Cells treated with GCA enhanced HAdV-induced cytopathic effects in epithelial and melanoma cancer cells but not normal cells, if the drug was applied several hours prior to HAdV inoculation. This was shown by real-time label-free impedance measurements using the xCELLigence system. GCA-treated cells contained fewer incoming HAdVs than control cells, but GCA treatment boosted HAdV titers and spreading in cancer cells. GCA enhanced viral gene expression or transgene expression from the cytomegalovirus promoter of B- or C-species HAdVs but did not enhance viral early region 1A (E1A) expression in uninfected cell lines or cells transfected with plasmid reporter DNA. The UPR-enhanced cell killing required the nuclease activity of the UPR sensor inositol-requiring enzyme 1 (IRE-1) and X box binding protein 1 (XBP-1), which alleviate ER stress. The collective results show that chemical UPR induction and viruses boost tumor cell killing by enhancing oncolytic viral efficacy.
Importance: Cancer is difficult to combat. A wide range of oncolytic viruses show promise for killing cancer cells, yet the efficacy of oncolytic killing is low. We searched for host factors enhancing adenovirus cancer cell killing and found that the knockdown of Golgi-specific brefeldin A-resistant guanine nucleotide exchange factor 1 (GBF-1) or chemical inhibition of GBF-1 enhanced adenovirus infection by triggering the IRE-1/XBP-1 branch of the unfolded protein response (UPR). IRE-1/XBP-1 promote cell survival and enhanced the levels of the adenoviral immediate early gene product E1A, virus spreading, and killing of cancer cells. Aggressive tumor cells depend on a readily inducible UPR and, hence, present prime targets for a combined strategy involving adenoviruses and small chemicals inducing UPR.
Copyright © 2014, American Society for Microbiology. All Rights Reserved.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
