

Hepatitis E virus of subtype 3i in chronically infected kidney transplant recipients in southeastern France
- PMID: 25187634
- PMCID: PMC4313232
- DOI: 10.1128/JCM.02028-14
Hepatitis E virus of subtype 3i in chronically infected kidney transplant recipients in southeastern France
Abstract
Hepatitis E virus (HEV) is a leading cause of waterborne acute hepatitis in developing countries. In Europe, HEV causes a zoonotic disease and is hyperendemic in southern France. Four HEV genotypes (1 to 4) have been defined, and the most used classification divides them into 24 subtypes. Autochthonous European HEV strains belong in majority to genotype 3. Subtypes 3c, 3f, and 3e are representative of the HEV diversity in France. HEV causes chronic hepatitis in solid-organ transplant recipients in Europe, and viral characteristics associated with chronicity are poorly documented. We sequenced 343-nucleotide-long HEV genomic fragments from the serum of eight chronically infected kidney transplant recipients and a near-full-length genome in one case. We identified in four patients (50%) HEV of subtype 3i, not described previously in France. If shorter genomic fragments were used in phylogenetic analyses, these HEV sequences were clustered with open reading frame 2 (ORF2) fragments labeled as subtype 3c. At least five of the eight HEV 3i sequences recovered from humans in our phylogenetic analyses were from chronically infected kidney transplant recipients. These data show that the description of the prevalence and geographical distribution of HEV subtypes may be partially inaccurate and that criteria for classification as 3i and 3c should be clarified. Extended molecular virology analyses are required to improve knowledge of HEV epidemiology and determinants of chronic HEV infection.
Copyright © 2014, American Society for Microbiology. All Rights Reserved.
Figures
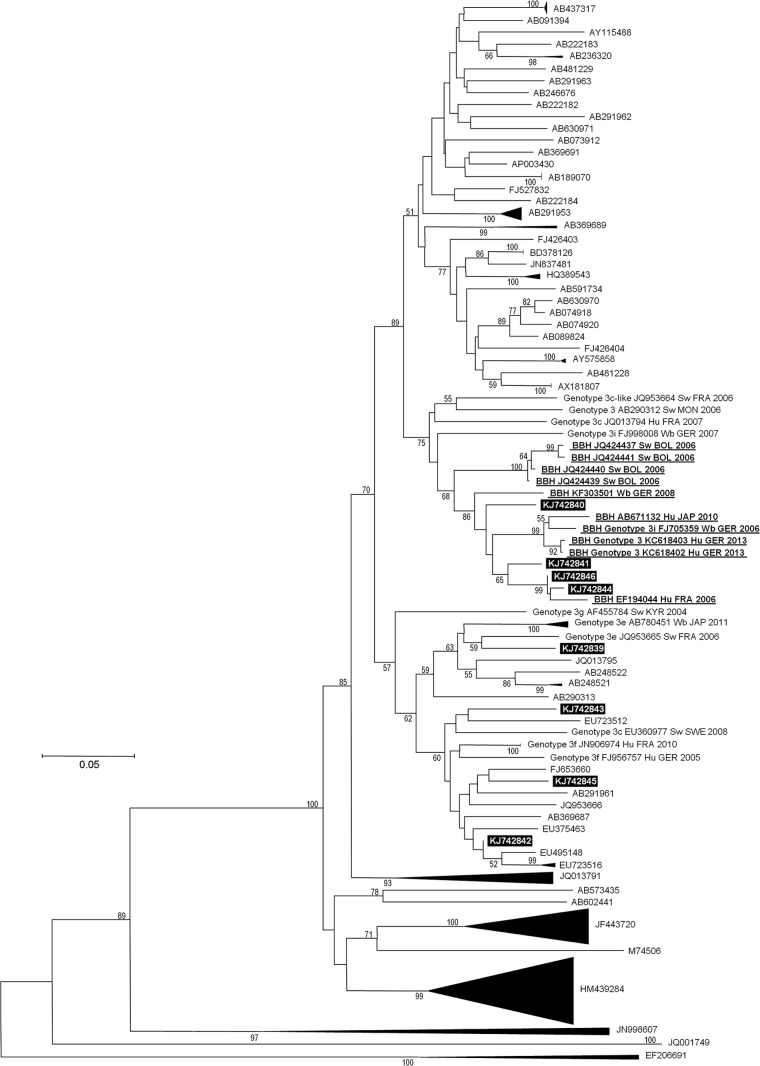
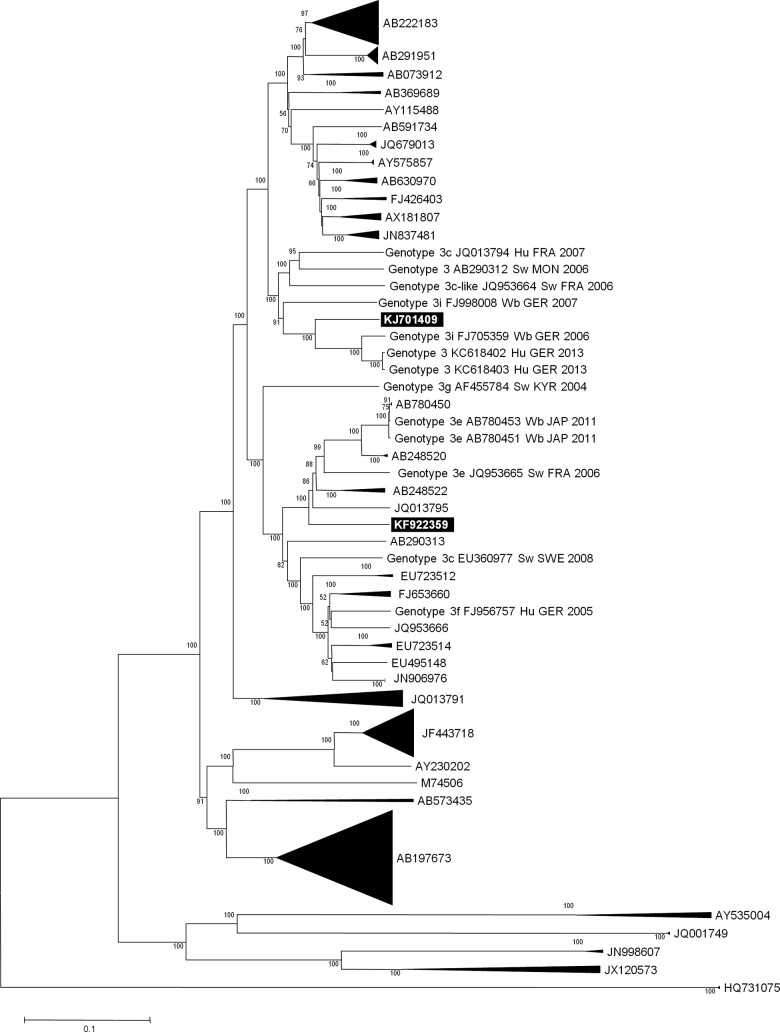
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
