

Structural basis for activation of trimeric Gi proteins by multiple growth factor receptors via GIV/Girdin
- PMID: 25187647
- PMCID: PMC4230624
- DOI: 10.1091/mbc.E14-05-0978
Structural basis for activation of trimeric Gi proteins by multiple growth factor receptors via GIV/Girdin
Abstract
A long-standing issue in the field of signal transduction is to understand the cross-talk between receptor tyrosine kinases (RTKs) and heterotrimeric G proteins, two major and distinct signaling hubs that control eukaryotic cell behavior. Although stimulation of many RTKs leads to activation of trimeric G proteins, the molecular mechanisms behind this phenomenon remain elusive. We discovered a unifying mechanism that allows GIV/Girdin, a bona fide metastasis-related protein and a guanine-nucleotide exchange factor (GEF) for Gαi, to serve as a direct platform for multiple RTKs to activate Gαi proteins. Using a combination of homology modeling, protein-protein interaction, and kinase assays, we demonstrate that a stretch of ∼110 amino acids within GIV C-terminus displays structural plasticity that allows folding into a SH2-like domain in the presence of phosphotyrosine ligands. Using protein-protein interaction assays, we demonstrated that both SH2 and GEF domains of GIV are required for the formation of a ligand-activated ternary complex between GIV, Gαi, and growth factor receptors and for activation of Gαi after growth factor stimulation. Expression of a SH2-deficient GIV mutant (Arg 1745→Leu) that cannot bind RTKs impaired all previously demonstrated functions of GIV-Akt enhancement, actin remodeling, and cell migration. The mechanistic and structural insights gained here shed light on the long-standing questions surrounding RTK/G protein cross-talk, set a novel paradigm, and characterize a unique pharmacological target for uncoupling GIV-dependent signaling downstream of multiple oncogenic RTKs.
© 2014 Lin, Ear, et al. This article is distributed by The American Society for Cell Biology under license from the author(s). Two months after publication it is available to the public under an Attribution–Noncommercial–Share Alike 3.0 Unported Creative Commons License (http://creativecommons.org/licenses/by-nc-sa/3.0).
Figures
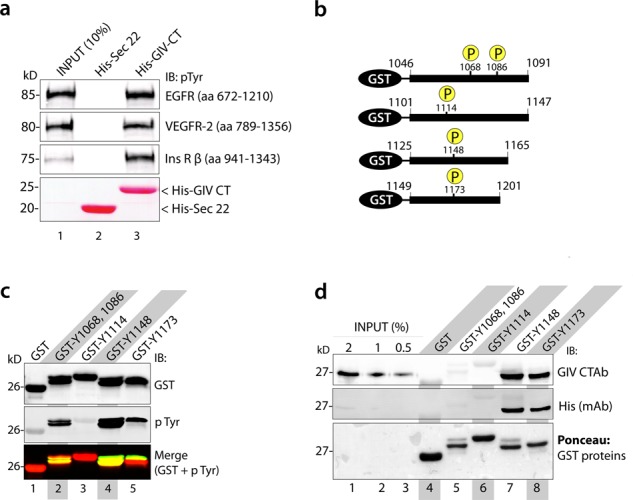

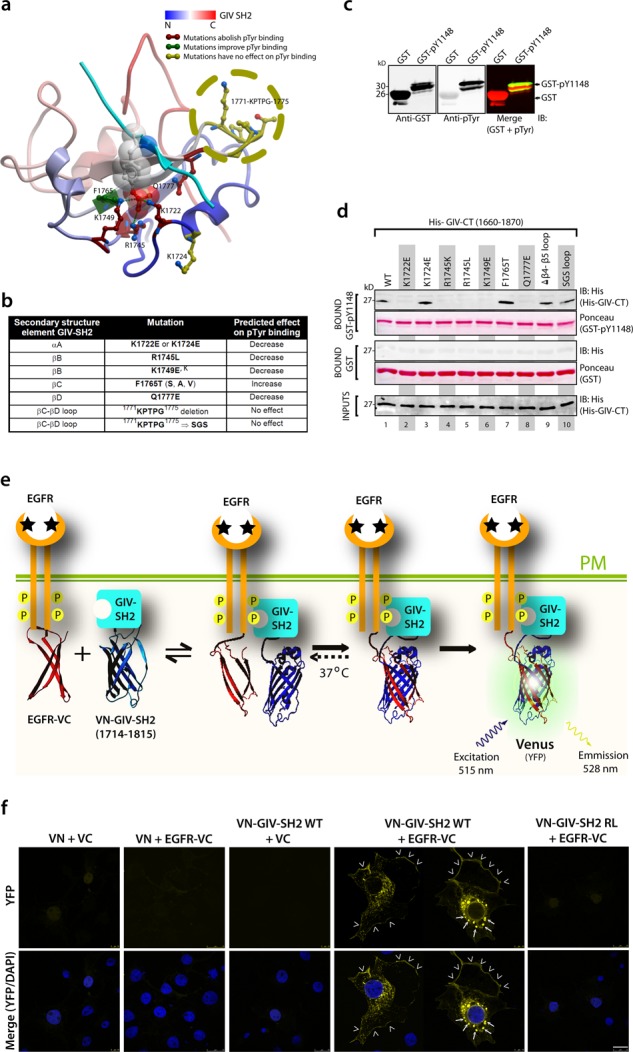





References
-
- Abagyan RA, Batalov S. Do aligned sequences share the same fold. J Mol Biol. 1997;273:355–368. - PubMed
-
- Abagyan R, Batalov S, Cardozo T, Totrov M, Webber J, Zhou Y. Homology modeling with internal coordinate mechanics: deformation zone mapping and improvements of models via conformational search. Proteins Suppl. 1997;1:29–37. - PubMed
-
- Abagyan R, Totrov M. Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J Mol Biol. 1994;235:983–1002. - PubMed
-
- Abagyan R, Totrov M, Kuznetsov DA. ICM: a new method for protein modeling and design: applications to docking and structure prediction from the distorted native conformation. J Comp Chem. 1994;15:488–506.
-
- Arold ST, Ulmer TS, Mulhern TD, Werner JM, Ladbury JE, Campbell ID, Noble ME. The role of the Src homology 3-Src homology 2 interface in the regulation of Src kinases. J Biol Chem. 2001;276:17199–17205. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
