

Discovering study-specific gene regulatory networks
- PMID: 25191999
- PMCID: PMC4156366
- DOI: 10.1371/journal.pone.0106524
Discovering study-specific gene regulatory networks
Abstract
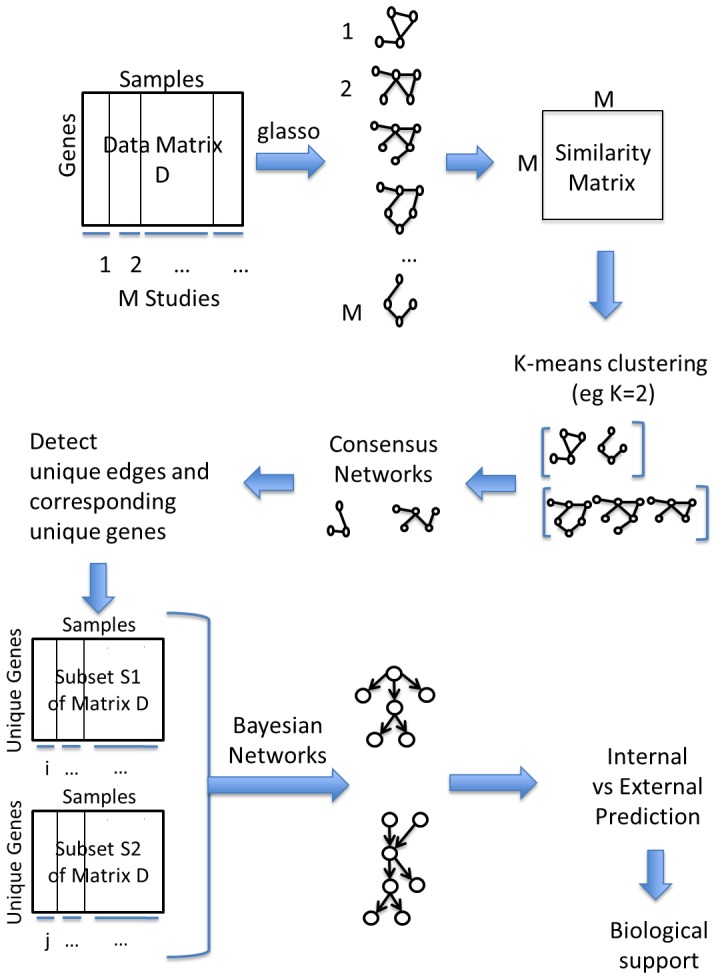
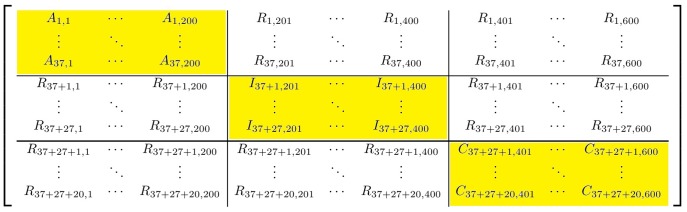
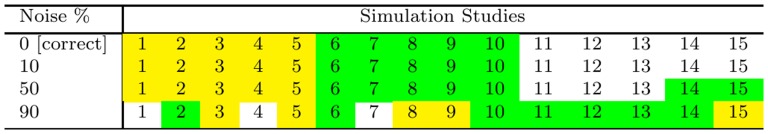
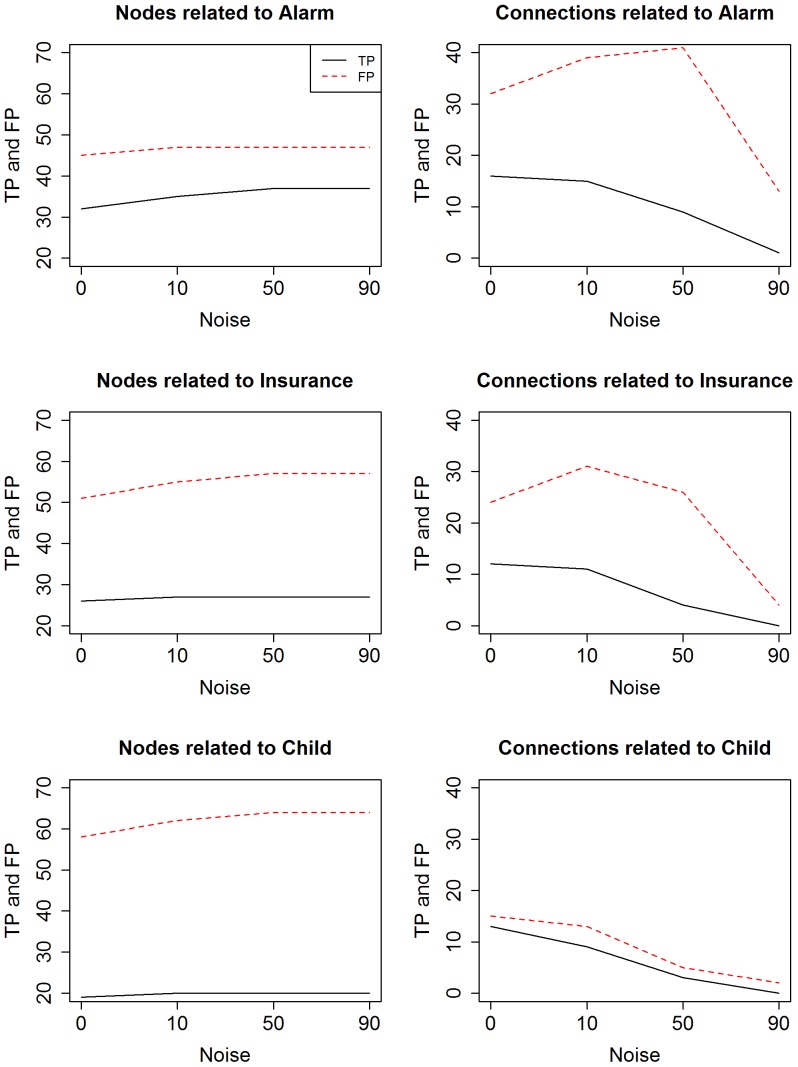
Microarrays are commonly used in biology because of their ability to simultaneously measure thousands of genes under different conditions. Due to their structure, typically containing a high amount of variables but far fewer samples, scalable network analysis techniques are often employed. In particular, consensus approaches have been recently used that combine multiple microarray studies in order to find networks that are more robust. The purpose of this paper, however, is to combine multiple microarray studies to automatically identify subnetworks that are distinctive to specific experimental conditions rather than common to them all. To better understand key regulatory mechanisms and how they change under different conditions, we derive unique networks from multiple independent networks built using glasso which goes beyond standard correlations. This involves calculating cluster prediction accuracies to detect the most predictive genes for a specific set of conditions. We differentiate between accuracies calculated using cross-validation within a selected cluster of studies (the intra prediction accuracy) and those calculated on a set of independent studies belonging to different study clusters (inter prediction accuracy). Finally, we compare our method's results to related state-of-the art techniques. We explore how the proposed pipeline performs on both synthetic data and real data (wheat and Fusarium). Our results show that subnetworks can be identified reliably that are specific to subsets of studies and that these networks reflect key mechanisms that are fundamental to the experimental conditions in each of those subsets.
Conflict of interest statement
Figures

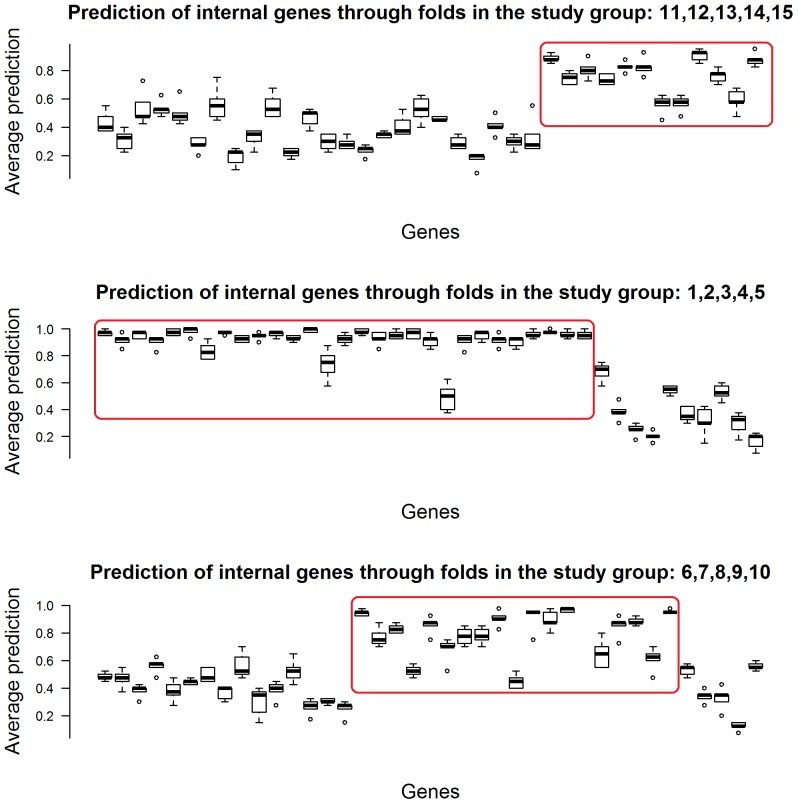
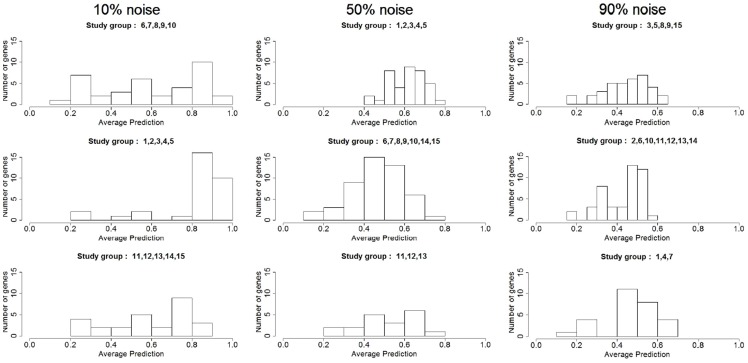
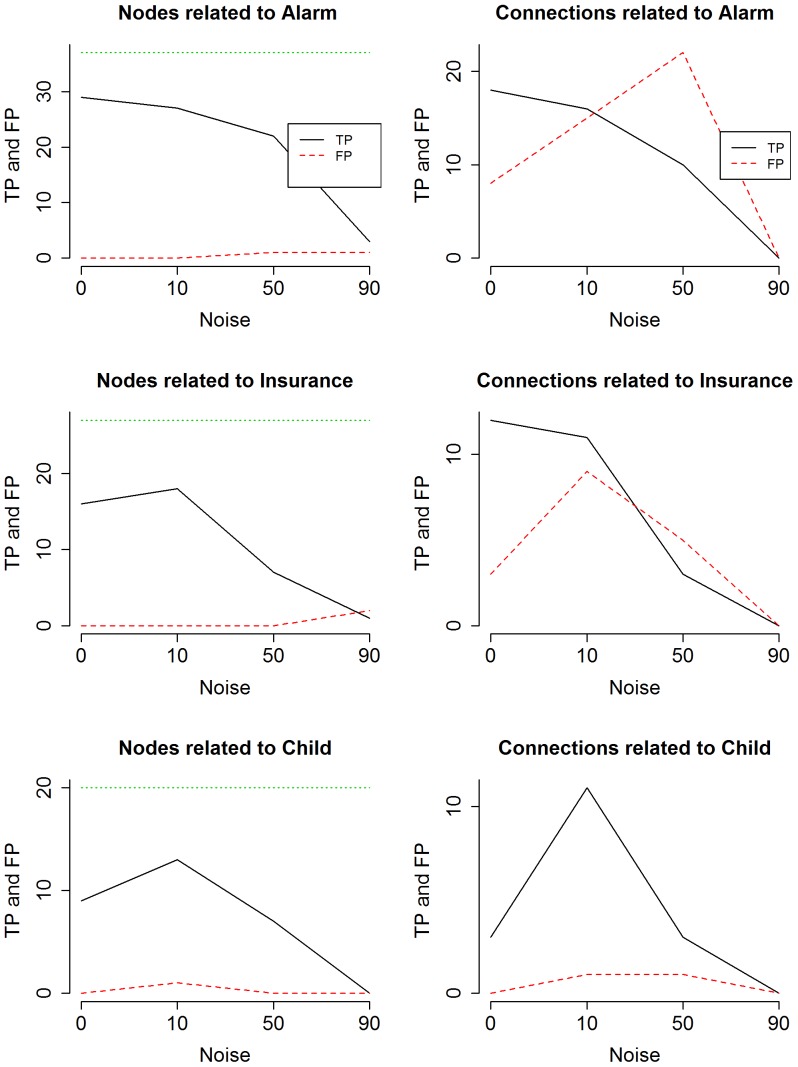
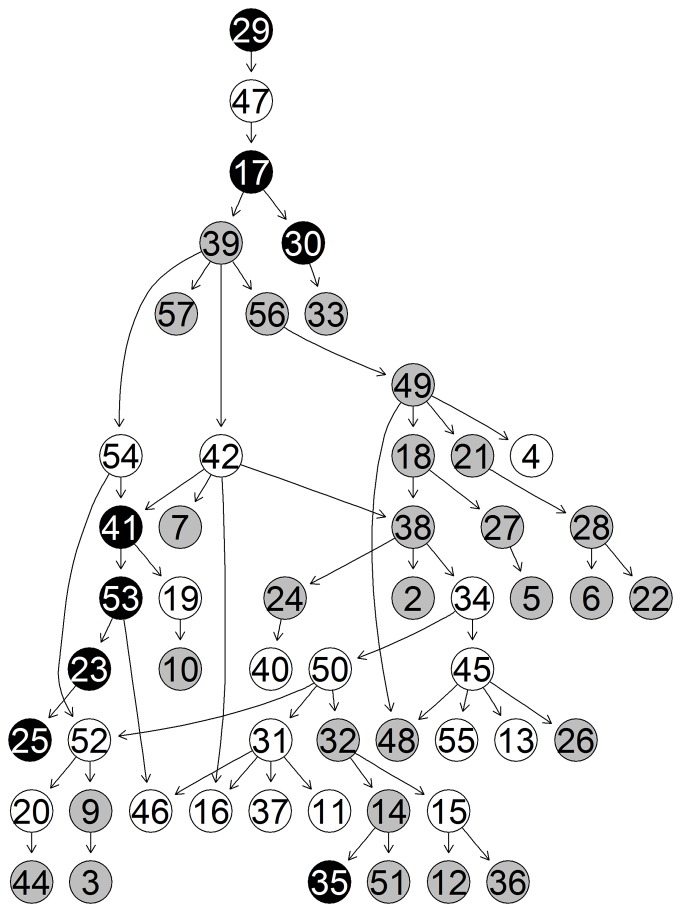
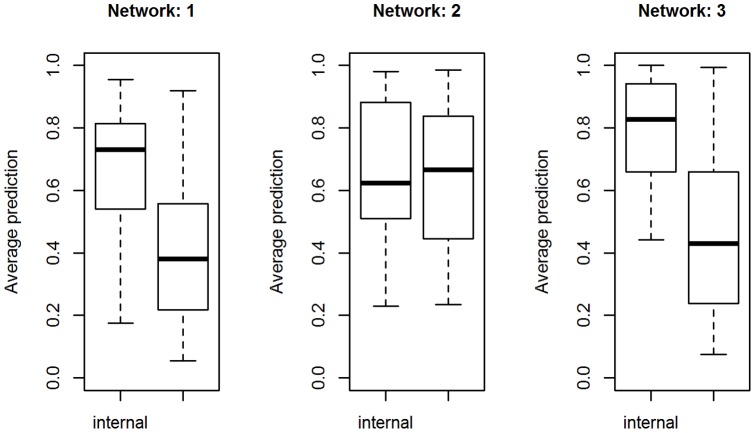
References
-
- Bo V, Lysenko A, Saqi M, Habash D, Tucker A (2013) Integrating multiple studies of wheat microarray data to identify treatment-specific regulatory networks. In: Advances in Intelligent Data Analysis XII, Springer. pp. 104–115.
-
- Choi J, Yu U, Kim S, Yoo O (2003) Combining multiple microarray studies and modeling interstudy variation. Bioinformatics 19: i84–i90. - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
