

Deep proteomic evaluation of primary and cell line motoneuron disease models delineates major differences in neuronal characteristics
- PMID: 25193168
- PMCID: PMC4256493
- DOI: 10.1074/mcp.M113.037291
Deep proteomic evaluation of primary and cell line motoneuron disease models delineates major differences in neuronal characteristics
Abstract
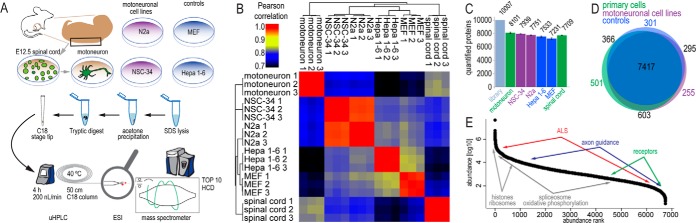
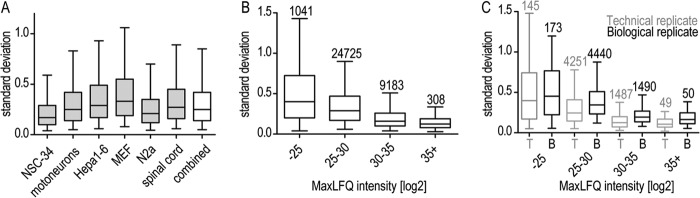
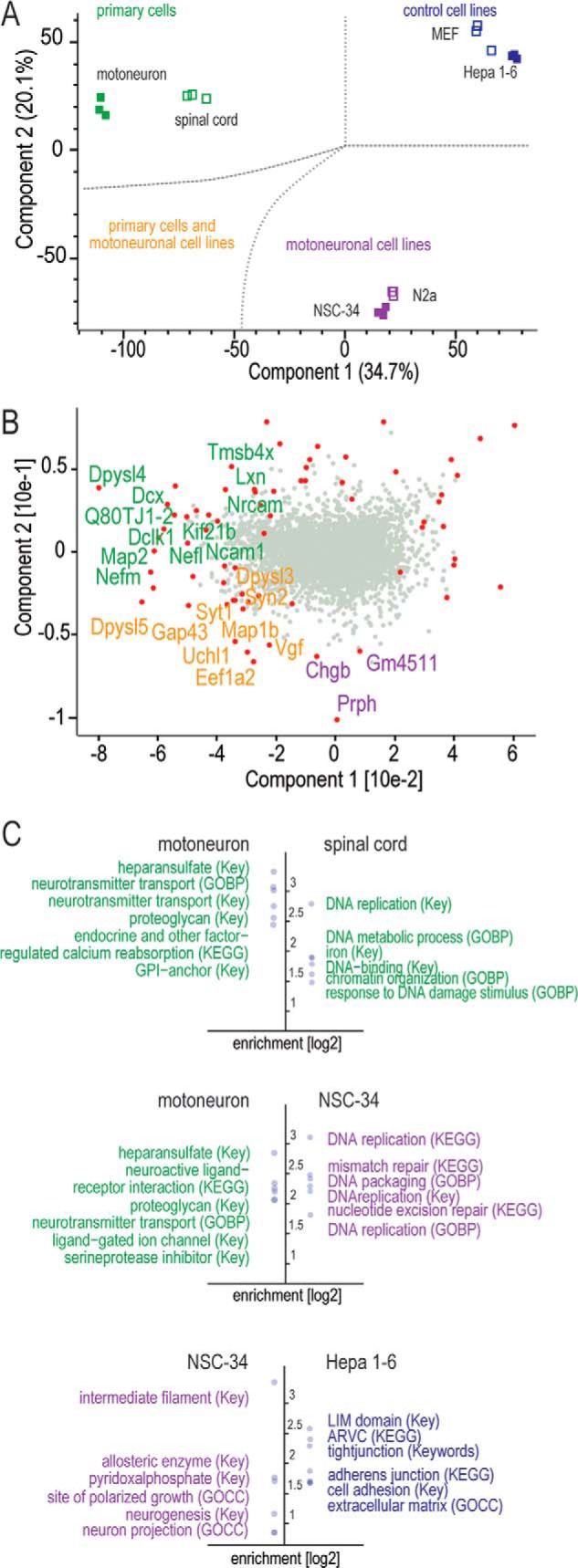
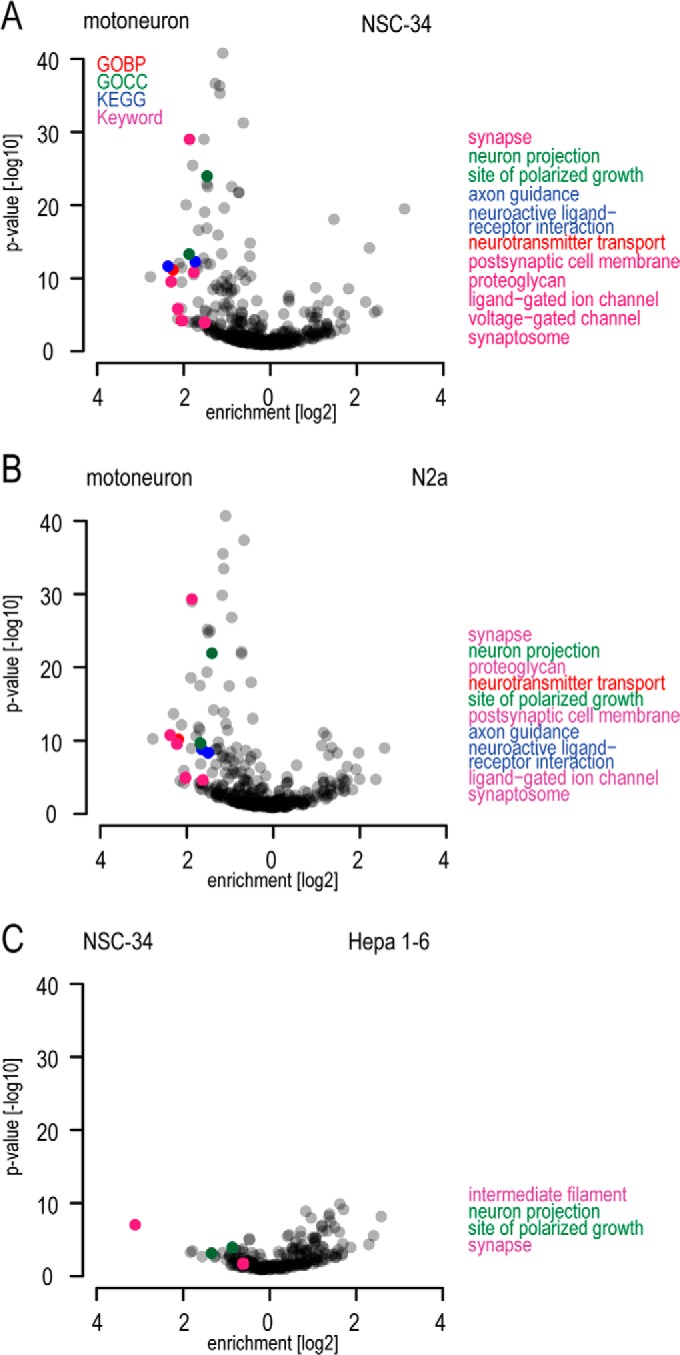
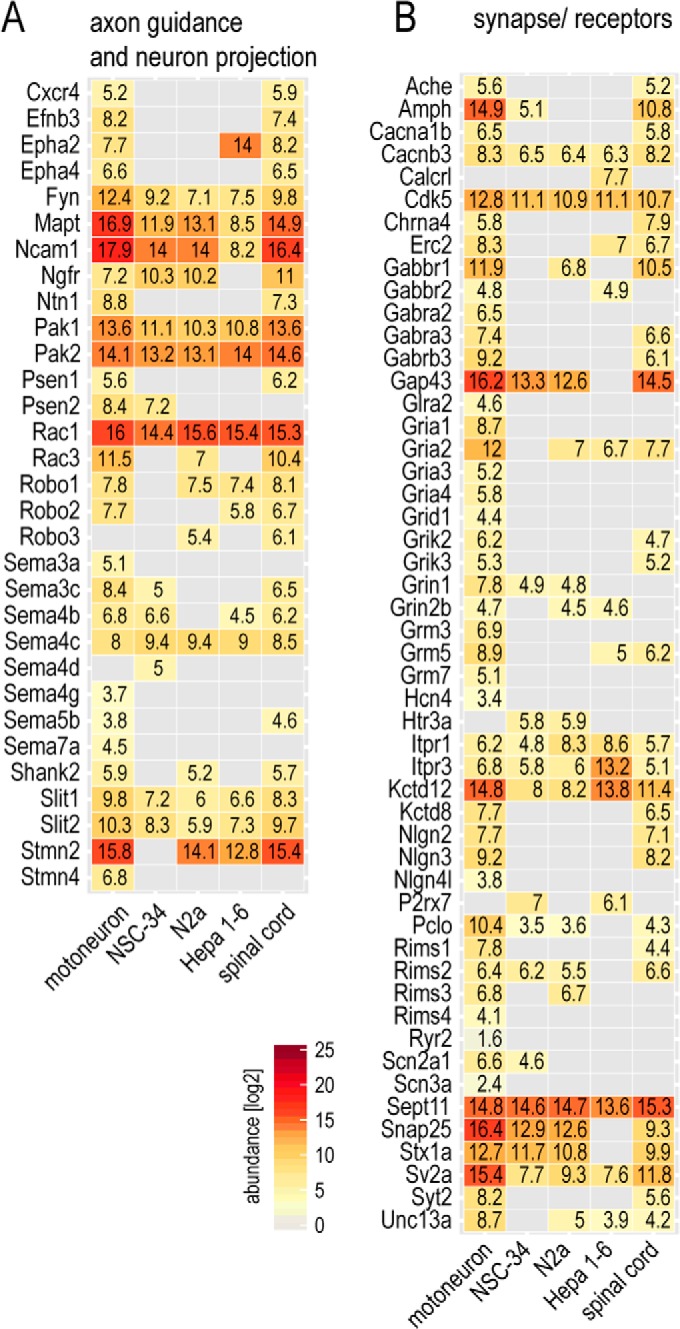
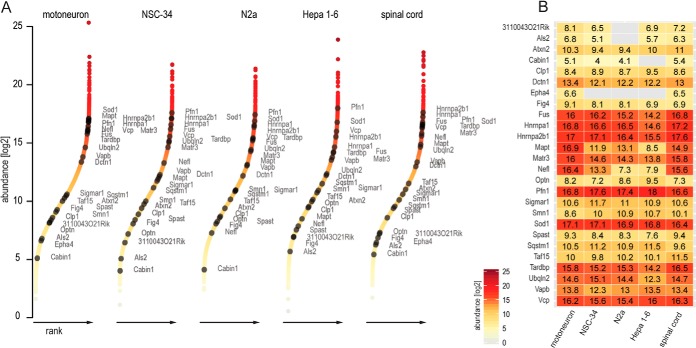
The fatal neurodegenerative disorders amyotrophic lateral sclerosis and spinal muscular atrophy are, respectively, the most common motoneuron disease and genetic cause of infant death. Various in vitro model systems have been established to investigate motoneuron disease mechanisms, in particular immortalized cell lines and primary neurons. Using quantitative mass-spectrometry-based proteomics, we compared the proteomes of primary motoneurons to motoneuron-like cell lines NSC-34 and N2a, as well as to non-neuronal control cells, at a depth of 10,000 proteins. We used this resource to evaluate the suitability of murine in vitro model systems for cell biological and biochemical analysis of motoneuron disease mechanisms. Individual protein and pathway analysis indicated substantial differences between motoneuron-like cell lines and primary motoneurons, especially for proteins involved in differentiation, cytoskeleton, and receptor signaling, whereas common metabolic pathways were more similar. The proteins associated with amyotrophic lateral sclerosis also showed distinct differences between cell lines and primary motoneurons, providing a molecular basis for understanding fundamental alterations between cell lines and neurons with respect to neuronal pathways with relevance for disease mechanisms. Our study provides a proteomics resource for motoneuron research and presents a paradigm of how mass-spectrometry-based proteomics can be used to evaluate disease model systems.
© 2014 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
References
-
- Lillo P., Hodges J. R. (2009) Frontotemporal dementia and motor neurone disease: overlapping clinic-pathological disorders. J. Clin. Neurosci. 16, 1131–1135 - PubMed
-
- Mackenzie I. R., Rademakers R., Neumann M. (2010) TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 9, 995–1007 - PubMed
-
- Rosen D. R. (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 364, 362. - PubMed
-
- Hadano S., Hand C. K., Osuga H., Yanagisawa Y., Otomo A., Devon R. S., Miyamoto N., Showguchi-Miyata J., Okada Y., Singaraja R., Figlewicz D. A., Kwiatkowski T., Hosler B. A., Sagie T., Skaug J., Nasir J., Brown R. H., Scherer S. W., Rouleau G. A., Hayden M. R., Ikeda J. E. (2001) A gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral sclerosis 2. Nat. Genet. 29, 166–173 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
