

Prolactin induces up-regulation of its cognate receptor in breast cancer cells via transcriptional activation of its generic promoter by cross-talk between ERα and STAT5
- PMID: 25193864
- PMCID: PMC4253420
- DOI: 10.18632/oncotarget.2376
Prolactin induces up-regulation of its cognate receptor in breast cancer cells via transcriptional activation of its generic promoter by cross-talk between ERα and STAT5
Abstract
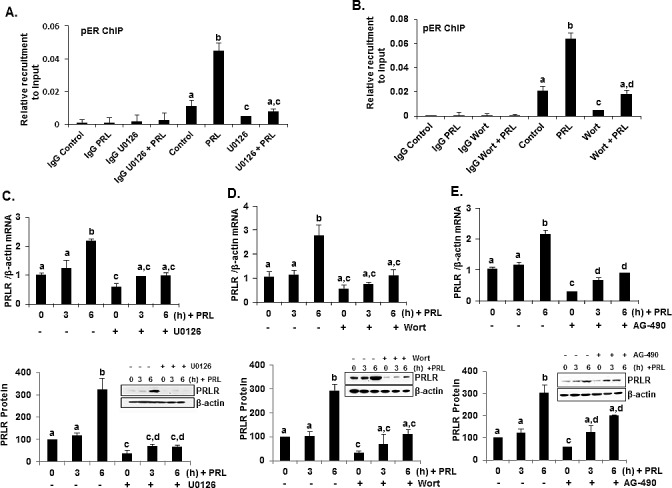
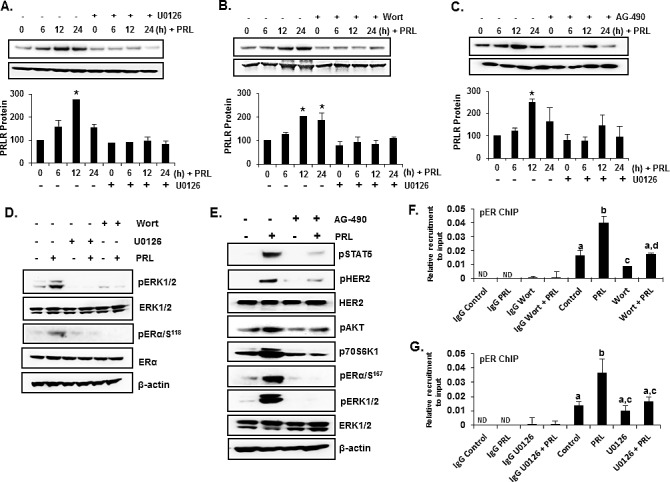
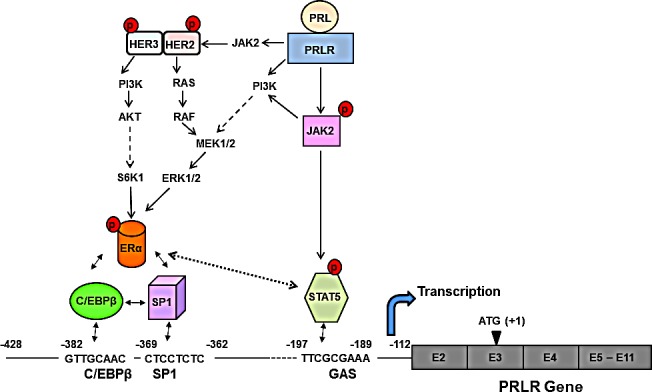
Prolactin (PRL) serves a critical role in breast cancer progression via activation of its cognate receptor. These studies reveal up-regulation of PRLR gene expression by PRL in absence of estradiol in MCF-7 and T47D breast cancer cells. PRL/PRLR via activation of STAT5 that binds a GAS-element in the PRLR gene and the participation of ERα stimulates PRLR transcription/expression. PRL/PRLR induces phosphorylation of ERα through the JAK2/PI3K/MAPK/ERK and JAK2/HER2 activated pathways. The increased recruitment of phospho-ERα, induced by PRL to Sp1 and C/EBPβ at PRLR promoter sites is essential for PRL-induced PRLR transcription. This recruitment is prevented by blockade of PRL expression using RNA interference or ERα phosphorylation using specific inhibitors of PI3K and ERK. Direct evidence is provided for local actions of PRL, independent of estradiol, in the up-regulation of PRLR transcription/expression by an activation-loop between STAT5 and the phospho-ERα/Sp1/C/EBPβ complex with requisite participation of signaling mechanisms. PRL's central role in the up-regulation of PRLR maximizes the action of the endogenous hormone. This study offers mechanistically rational basis for invasiveness fueled by prolactin in refractory states to adjuvant therapies in breast cancer.
Figures
References
-
- Welsch CW, Nagasawa H. Prolactin and murine mammary tumorigenesis: a review. Cancer Res. 1977;37:951–963. - PubMed
-
- Wennbo H, Tornell J. The role of prolactin and growth hormone in breast cancer. Oncogene. 2000;19:1072–1076. - PubMed
-
- Bonneterre J, Peyrat JP, Beuscart R, Demaille A. Biological and clinical aspects of prolactin receptors (PRL-R) in human breast cancer. J Steroid Biochem Mol Biol. 1990;37:977–981. - PubMed
-
- Harvey PW. Hypothesis: prolactin is tumorigenic to human breast: dispelling the myth that prolactin-induced mammary tumors are rodent-specific. J App Toxicol. 2012;32:1–9. - PubMed
-
- Hu ZZ, Meng J, Dufau ML. Isolation and characterization of two novel forms of the human prolactin receptor generated by alternative splicing of a newly identified exon 11. J Biol Chem. 2001;276:41086–41094. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
