

Clinical and genetical heterogeneity of late-onset multiple acyl-coenzyme A dehydrogenase deficiency
- PMID: 25200064
- PMCID: PMC4222585
- DOI: 10.1186/s13023-014-0117-5
Clinical and genetical heterogeneity of late-onset multiple acyl-coenzyme A dehydrogenase deficiency
Abstract
Background: Multiple acyl-CoA dehydrogenase deficiency (MADD) is an autosomal recessive disorder caused by deficiency of electron transfer flavoprotein or electron transfer flavoprotein dehydrogenase. The clinical picture of late-onset forms is highly variable with symptoms ranging from acute metabolic decompensations to chronic, mainly muscular problems or even asymptomatic cases.
Methods: All 350 cases of late-onset MADD reported in the literature to date have been analyzed and evaluated with respect to age at presentation, diagnostic delay, biochemical features and diagnostic parameters as well as response to treatment.
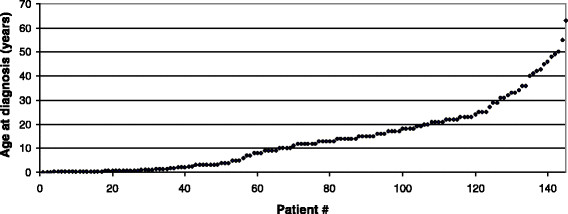
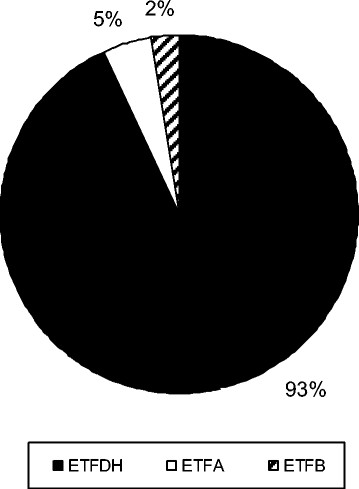
Results: Mean age at onset was 19.2 years. The mean delay between onset of symptoms and diagnosis was 3.9 years. Chronic muscular symptoms were more than twice as common as acute metabolic decompensations (85% versus 33% of patients, respectively). 20% had both acute and chronic symptoms. 5% of patients had died at a mean age of 5.8 years, while 3% of patients have remained asymptomatic until a maximum age of 14 years. Diagnosis may be difficult as a relevant number of patients do not display typical biochemical patterns of urine organic acids and blood acylcarnitines during times of wellbeing. The vast majority of patients carry mutations in the ETFDH gene (93%), while mutations in the ETFA (5%) and ETFB (2%) genes are the exceptions. Almost all patients with late-onset MADD (98%) are clearly responsive to riboflavin.
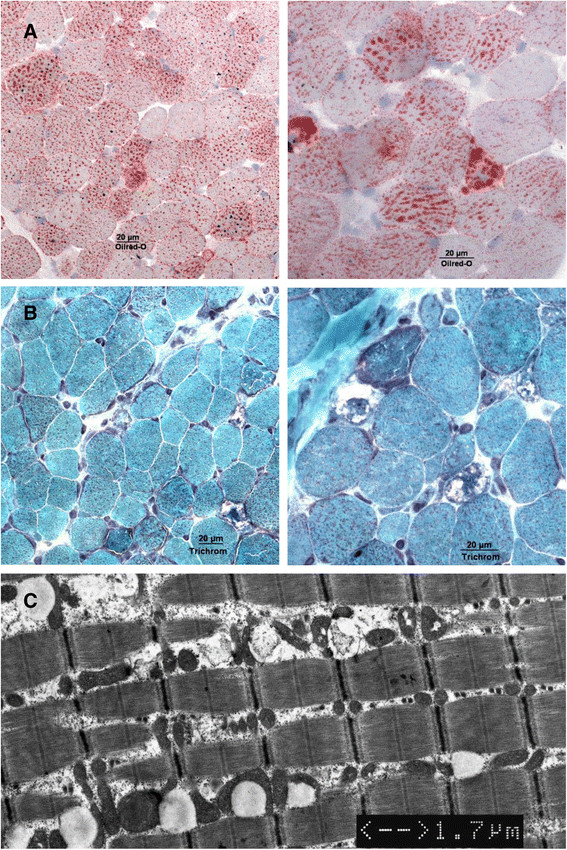
Conclusions: Late-onset MADD is probably an underdiagnosed disease and should be considered in all patients with acute or chronic muscular symptoms or acute metabolic decompensation with hypoglycemia, acidosis, encephalopathy and hepatopathy. This may not only prevent patients from invasive diagnostic procedures such as muscle biopsies, but also help to avoid fatal metabolic decompensations.
Figures
References
-
- Frerman FE, Goodman SI. In: The Metabolic and Molecular Bases of Inherited Disease. Scriver CR, Sly WS, Childs B, Beaudet AL, Valle D, Kinzler KW, Vogelstein B, editor. 2001. Defects of Electron Transfer Flavoprotein and Electron Transfer Flavoprotein-Ubiquinone Oxidoreductase: Glutaric Acidemia Type II.
-
- Goodman SI, Frerman FE. Glutaric acidaemia type II (multiple acyl-CoA dehydrogenation deficiency) J Inherit Metab Dis. 1984;7(Suppl 1):33–37. - PubMed
-
- Dusheiko G, Kew MC, Joffe BI, Lewin JR, Mantagos S, Tanaka K. Recurrent hypoglycemia associated with glutaric aciduria type II in an adult. N Engl J Med. 1979;301(26):1405–1409. - PubMed
-
- Bell RB, Brownell AK, Roe CR, Engel AG, Goodman SI, Frerman FE, Seccombe DW, Snyder FF. Electron transfer flavoprotein: ubiquinone oxidoreductase (ETF:QO) deficiency in an adult. Neurology. 1990;40(11):1779–1782. - PubMed
-
- Izumi R, Suzuki N, Nagata M, Hasegawa T, Abe Y, Saito Y, Mochizuki H, Tateyama M, Aoki M. A case of late onset riboflavin-responsive multiple acyl-CoA dehydrogenase deficiency manifesting as recurrent rhabdomyolysis and acute renal failure. Intern Med. 2011;50(21):2663–2668. - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
