

Involvement of the host DNA-repair enzyme TDP2 in formation of the covalently closed circular DNA persistence reservoir of hepatitis B viruses
- PMID: 25201958
- PMCID: PMC4209993
- DOI: 10.1073/pnas.1409986111
Involvement of the host DNA-repair enzyme TDP2 in formation of the covalently closed circular DNA persistence reservoir of hepatitis B viruses
Abstract
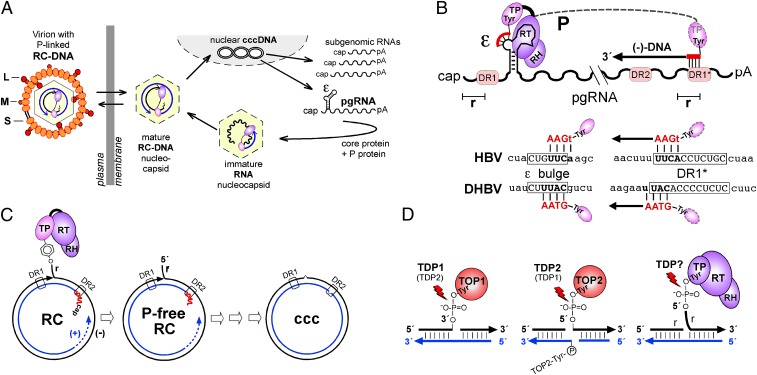
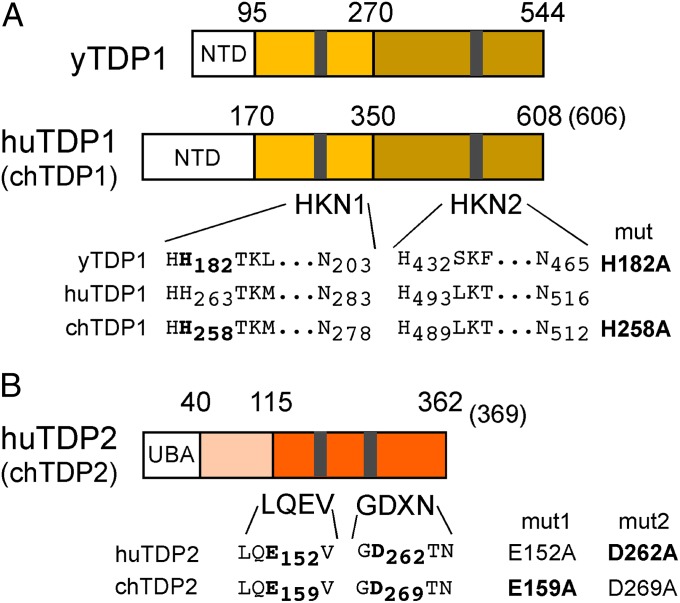
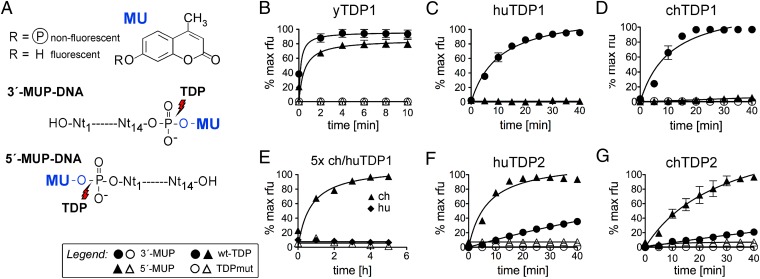
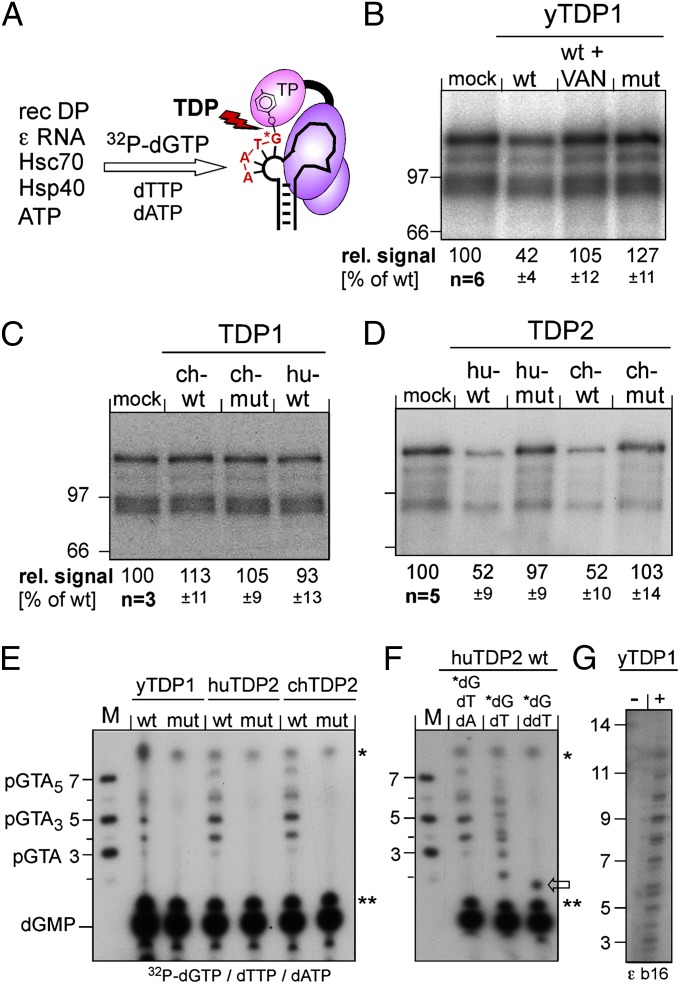
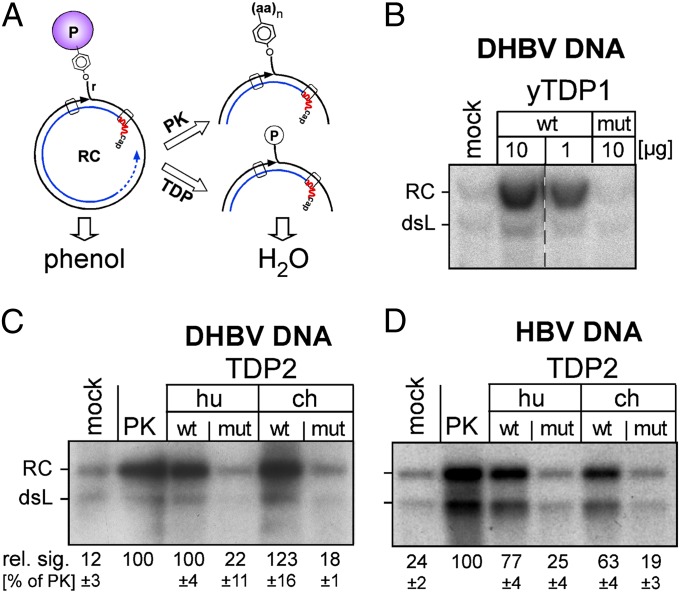
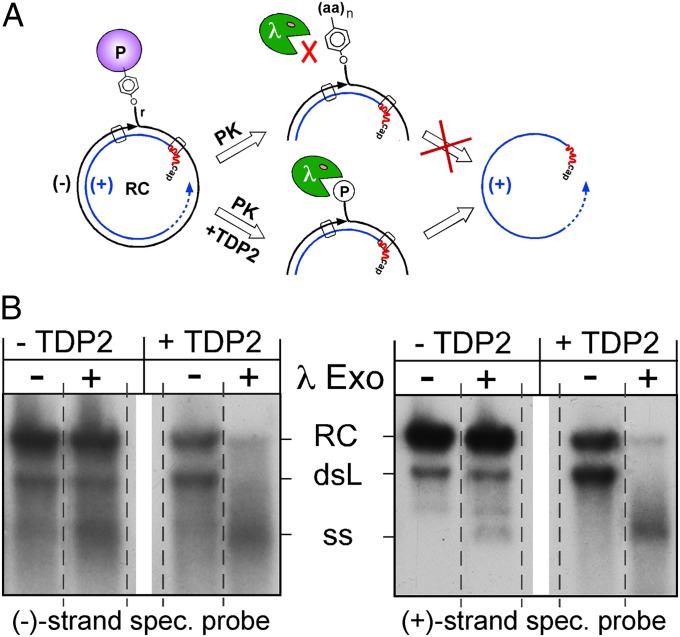
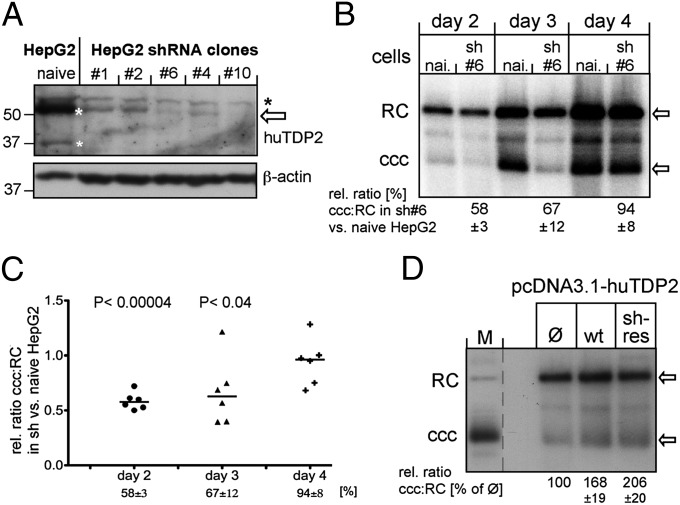
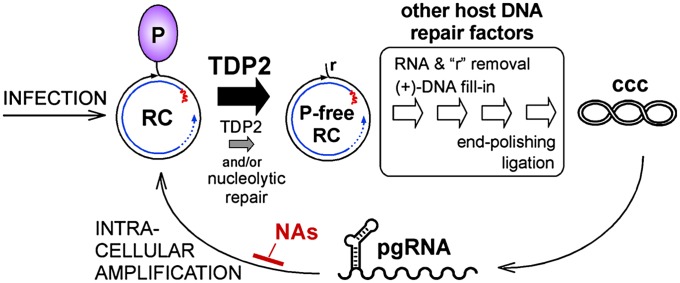
Hepatitis B virus (HBV), the causative agent of chronic hepatitis B and prototypic hepadnavirus, is a small DNA virus that replicates by protein-primed reverse transcription. The product is a 3-kb relaxed circular DNA (RC-DNA) in which one strand is linked to the viral polymerase (P protein) through a tyrosyl-DNA phosphodiester bond. Upon infection, the incoming RC-DNA is converted into covalently closed circular (ccc) DNA, which serves as a viral persistence reservoir that is refractory to current anti-HBV treatments. The mechanism of cccDNA formation is unknown, but the release of P protein is one mandatory step. Structural similarities between RC-DNA and cellular topoisomerase-DNA adducts and their known repair by tyrosyl-DNA-phosphodiesterase (TDP) 1 or TDP2 suggested that HBV may usurp these enzymes for its own purpose. Here we demonstrate that human and chicken TDP2, but only the yeast ortholog of TDP1, can specifically cleave the Tyr-DNA bond in virus-adapted model substrates and release P protein from authentic HBV and duck HBV (DHBV) RC-DNA in vitro, without prior proteolysis of the large P proteins. Consistent with TPD2's having a physiological role in cccDNA formation, RNAi-mediated TDP2 depletion in human cells significantly slowed the conversion of RC-DNA to cccDNA. Ectopic TDP2 expression in the same cells restored faster conversion kinetics. These data strongly suggest that TDP2 is a first, although likely not the only, host DNA-repair factor involved in HBV cccDNA biogenesis. In addition to establishing a functional link between hepadnaviruses and DNA repair, our results open new prospects for directly targeting HBV persistence.
Keywords: TDP substrate specificity; hepatitis B virus persistence; virus–DNA repair interface.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Ott JJ, Stevens GA, Groeger J, Wiersma ST. Global epidemiology of hepatitis B virus infection: New estimates of age-specific HBsAg seroprevalence and endemicity. Vaccine. 2012;30(12):2212–2219. - PubMed
-
- Ganem D, Prince AM. Hepatitis B virus infection—natural history and clinical consequences. N Engl J Med. 2004;350(11):1118–1129. - PubMed
-
- Zoulim F, Locarnini S. Hepatitis B virus resistance to nucleos(t)ide analogues. Gastroenterology. 2009;137(5):1593–1608 e1591-1592. - PubMed
-
- Scaglione SJ, Lok AS. Effectiveness of hepatitis B treatment in clinical practice. Gastroenterology. 2012;142(6):1360–1368 e1361. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
