

MicroRNAs 206 and 21 cooperate to promote RAS-extracellular signal-regulated kinase signaling by suppressing the translation of RASA1 and SPRED1
- PMID: 25202123
- PMCID: PMC4248710
- DOI: 10.1128/MCB.00480-14
MicroRNAs 206 and 21 cooperate to promote RAS-extracellular signal-regulated kinase signaling by suppressing the translation of RASA1 and SPRED1
Abstract
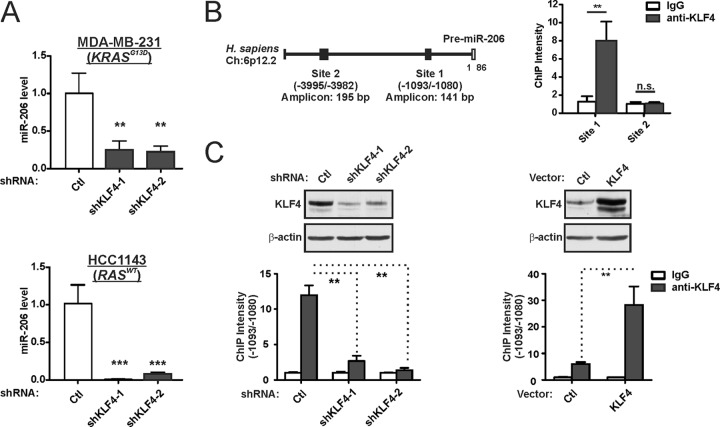
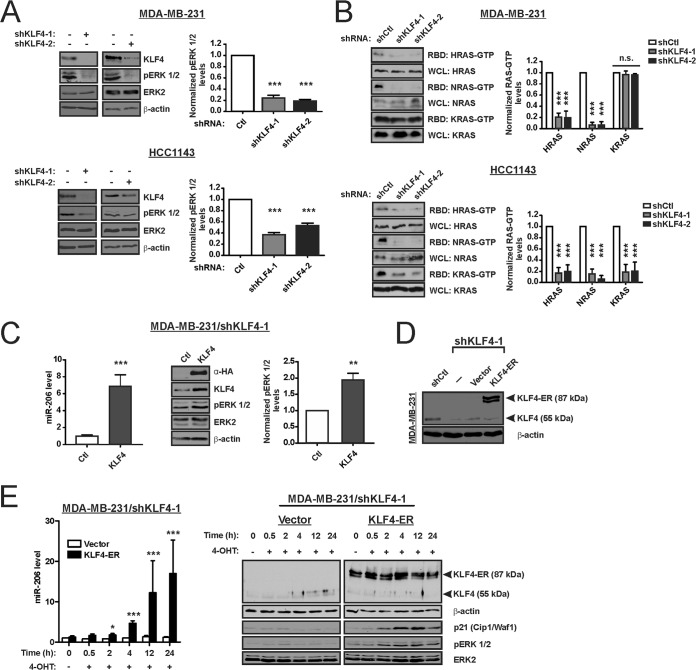
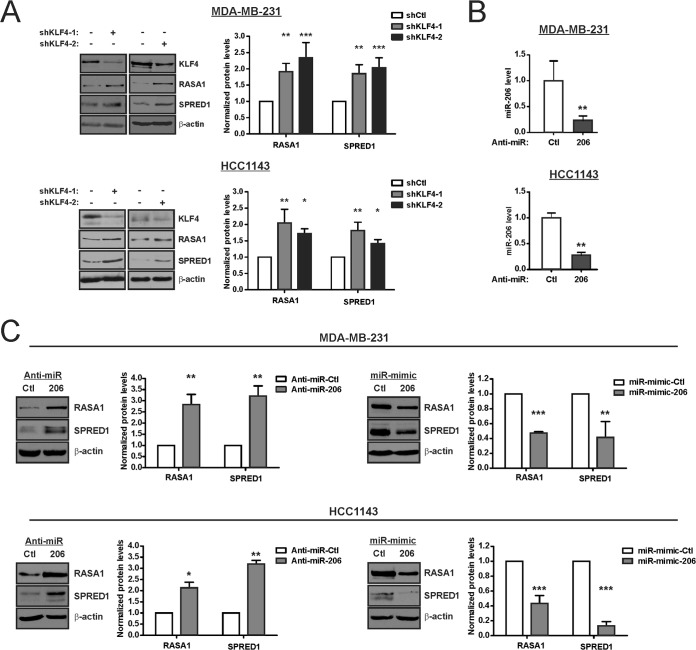
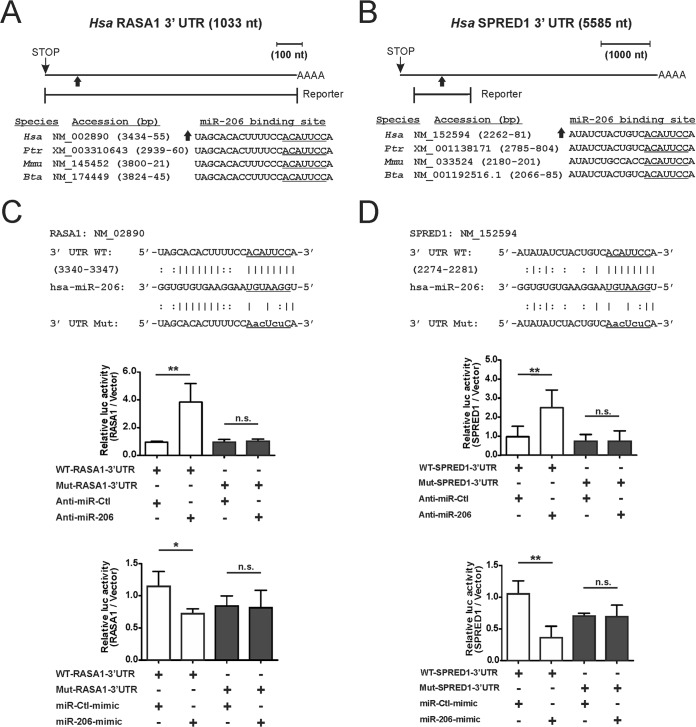
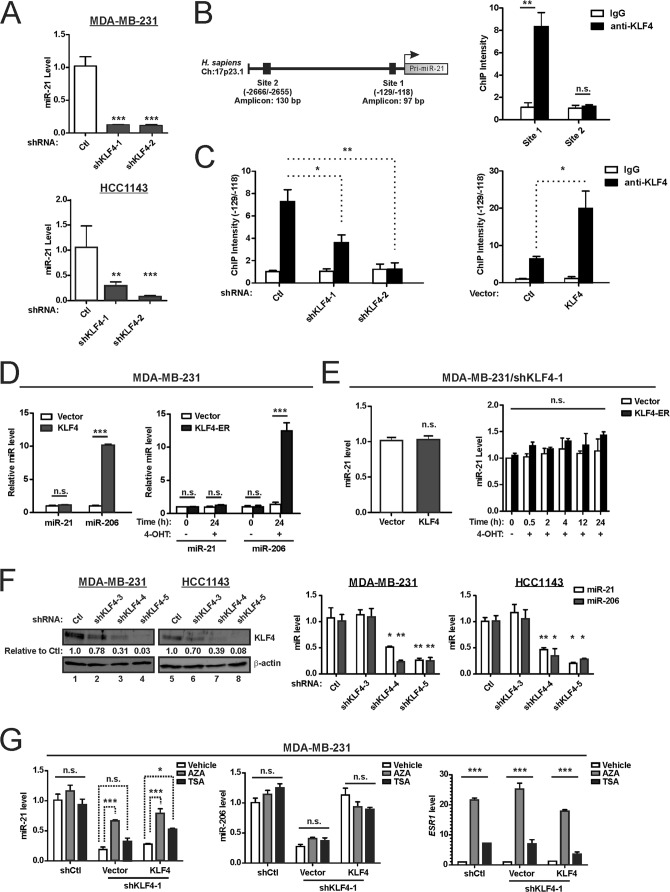
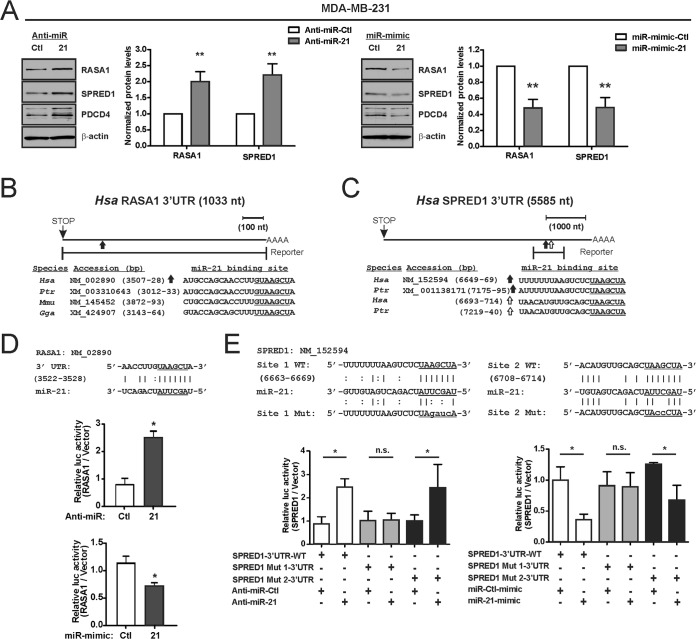
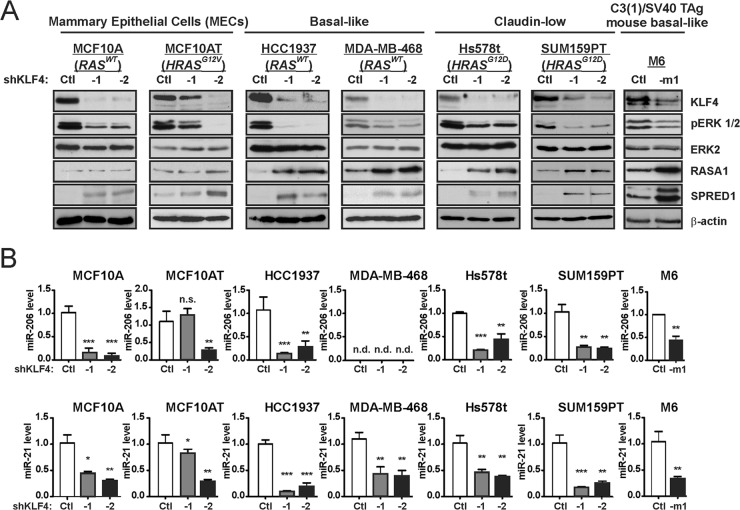
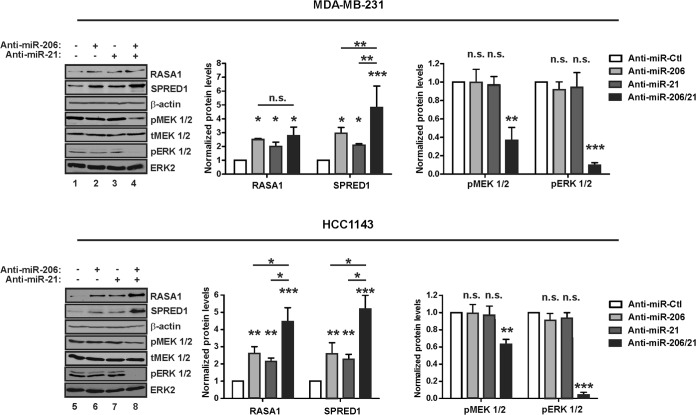
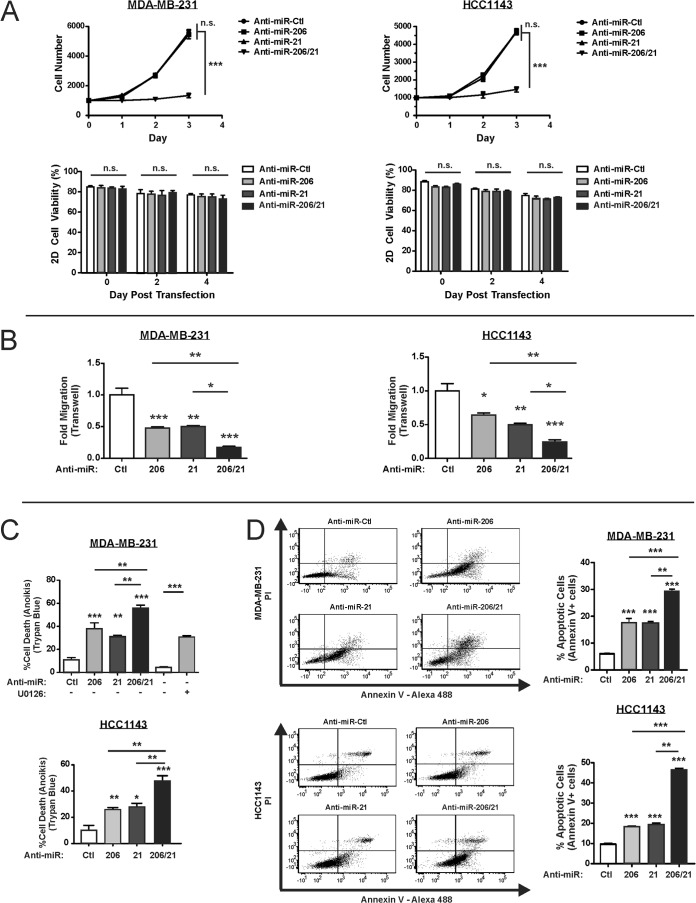
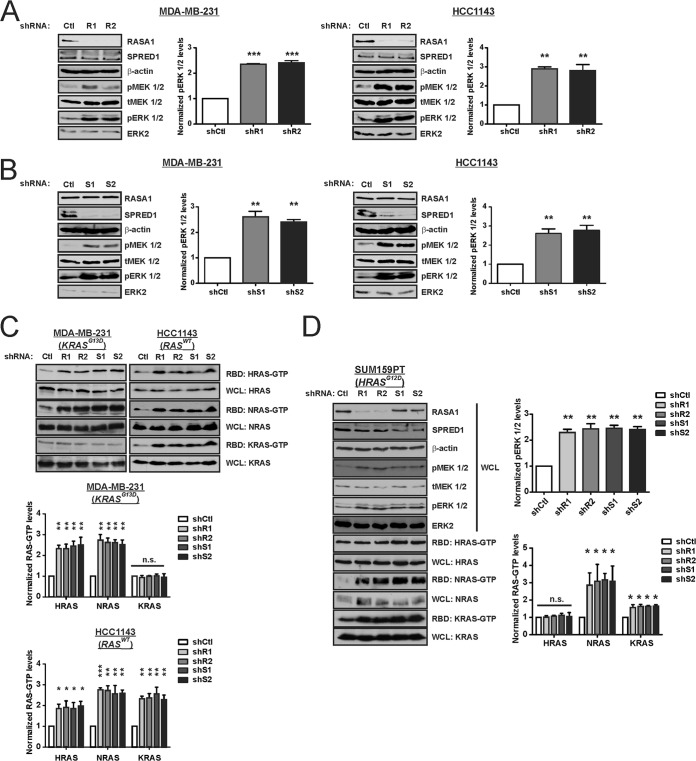
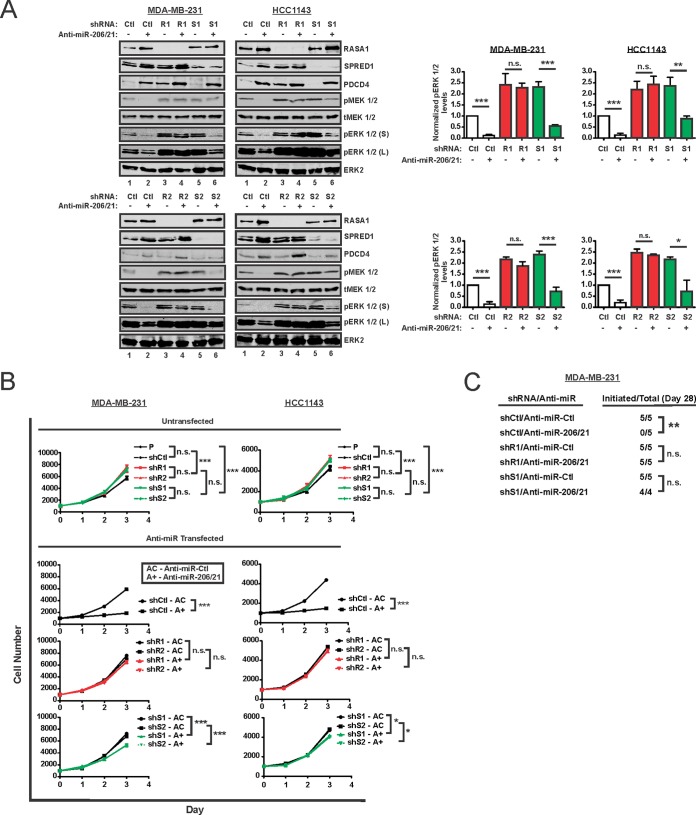
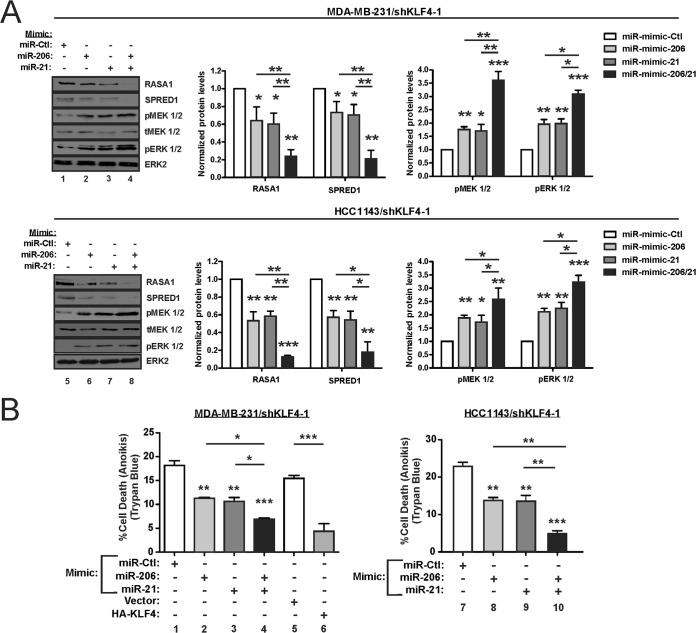
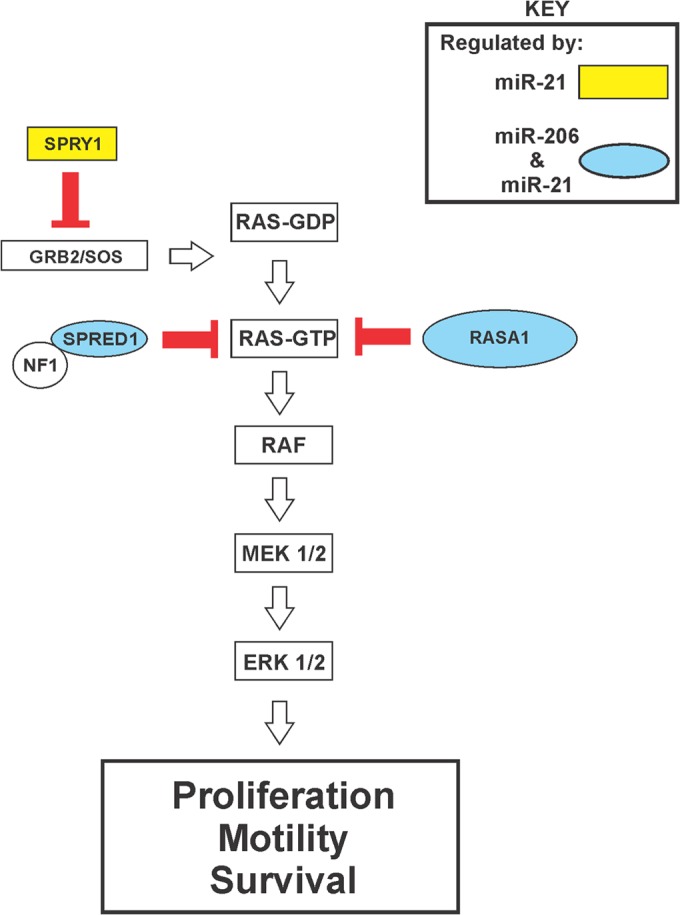
Despite the low prevalence of activating point mutation of RAS or RAF genes, the RAS-extracellular signal-regulated kinase (ERK) pathway is implicated in breast cancer pathogenesis. Indeed, in triple-negative breast cancer (TNBC), there is recurrent genetic alteration of pathway components. Using short hairpin RNA (shRNA) methods, we observed that the zinc finger transcription factor Krüppel-like factor 4 (KLF4) can promote RAS-ERK signaling in TNBC cells. Endogenous KLF4 bound to the promoter regions and promoted the expression of two microRNAs (miRs), miR-206 and miR-21 (i.e., miR-206/21). Antisense-mediated knockdown (anti-miR) revealed that miR-206/21 coordinately promote RAS-ERK signaling and the corresponding cell phenotypes by inhibiting translation of the pathway suppressors RASA1 and SPRED1. In TNBC cells, including cells with mutation of RAS, the suppression of either RASA1 or SPRED1 increased the levels of GTP-bound, wild-type RAS and activated ERK 1/2. Unlike the control cells, treatment of RASA1- or SPRED1-suppressed cells with anti-miR-206/21 had little or no impact on the level of activated ERK 1/2 or on cell proliferation and failed to suppress tumor initiation. These results identify RASA1 and SPRED1 mRNAs as latent RAS-ERK pathway suppressors that can be upregulated in tumor cells by anti-miR treatment. Consequently, KLF4-regulated miRs are important for the maintenance of RAS-ERK pathway activity in TNBC cells.
Copyright © 2014, American Society for Microbiology. All Rights Reserved.
Figures
References
-
- Weinberg RA. 2007. Growth factors, receptors, and cancer, p 119–158 In Weinberg RA. (ed), The biology of cancer. Garland Science, Taylor and Francis Group, LLC, New York, NY.
-
- McCubrey JA, Steelman LS, Abrams SL, Lee JT, Chang F, Bertrand FE, Navolanic PM, Terrian DM, Franklin RA, D'Assoro AB, Salisbury JL, Mazzarino MC, Stivala F, Libra M. 2006. Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT pathways in malignant transformation and drug resistance. Adv. Enzyme Regul. 46:249–279. 10.1016/j.advenzreg.2006.01.004. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous