

Oxidative stress, mitochondrial damage and neurodegenerative diseases
- PMID: 25206509
- PMCID: PMC4145906
- DOI: 10.3969/j.issn.1673-5374.2013.21.009
Oxidative stress, mitochondrial damage and neurodegenerative diseases
Abstract
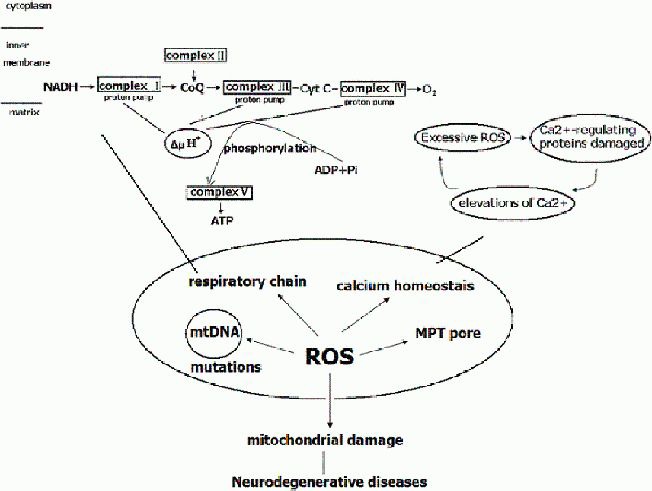
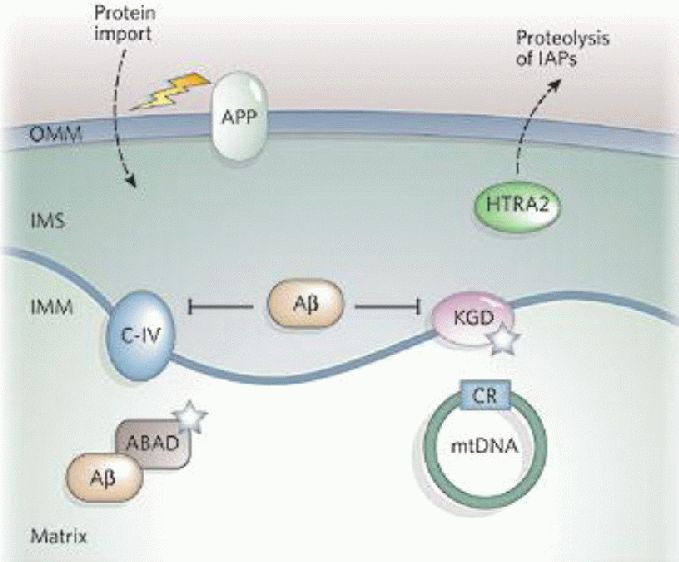
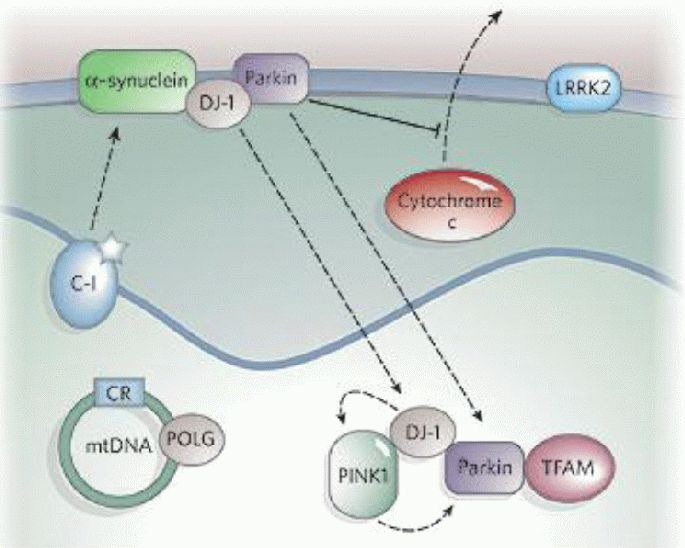
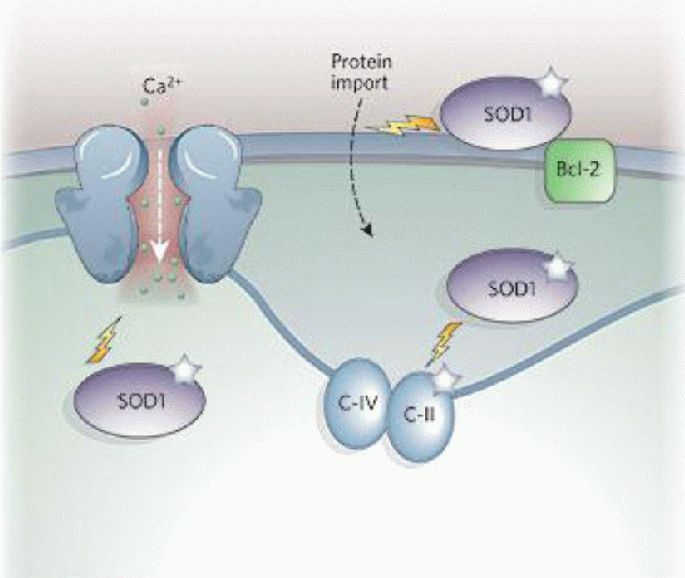
Oxidative stress and mitochondrial damage have been implicated in the pathogenesis of several neurodegenerative diseases, including Alzheimer's disease, Parkinson's disease and amyotrophic lateral sclerosis. Oxidative stress is characterized by the overproduction of reactive oxygen species, which can induce mitochondrial DNA mutations, damage the mitochondrial respiratory chain, alter membrane permeability, and influence Ca(2+) homeostasis and mitochondrial defense systems. All these changes are implicated in the development of these neurodegenerative diseases, mediating or amplifying neuronal dysfunction and triggering neurodegeneration. This paper summarizes the contribution of oxidative stress and mitochondrial damage to the onset of neurodegenerative eases and discusses strategies to modify mitochondrial dysfunction that may be attractive therapeutic interventions for the treatment of various neurodegenerative diseases.
Keywords: Alzheimer's disease; Parkinson's disease; amyotrophic lateral sclerosis; grants-supported paper; mitochondrial damage; neural regeneration; neurodegenerative diseases; neuroregeneration; oxidative stress; reactive oxygen species; respiratory chain.
Conflict of interest statement
Figures
References
-
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443(7113):787–795. - PubMed
-
- Crunkhorn S. Neurodegenerative disorders: Restoring the balance. Nat Rev Drug Discov. 2011;10(8):576. - PubMed
-
- Sas K, Robotka H, Toldi J, et al. Mitochondria, metabolic disturbances, oxidative stress and the kynurenine system, with focus on neurodegenerative disorders. J Neurol Sci. 2007;257(1-2):221–239. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous