

Fast and accurate discovery of degenerate linear motifs in protein sequences
- PMID: 25207816
- PMCID: PMC4160167
- DOI: 10.1371/journal.pone.0106081
Fast and accurate discovery of degenerate linear motifs in protein sequences
Abstract
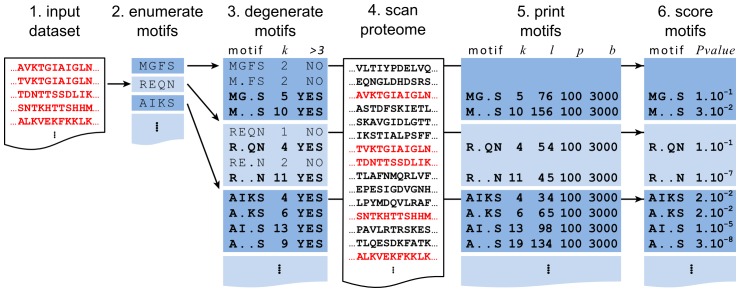
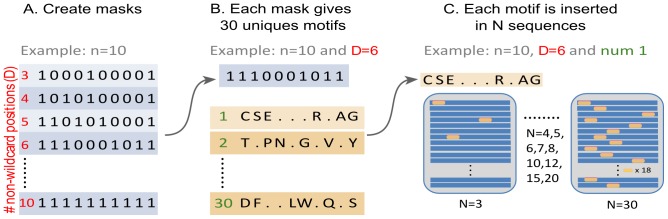
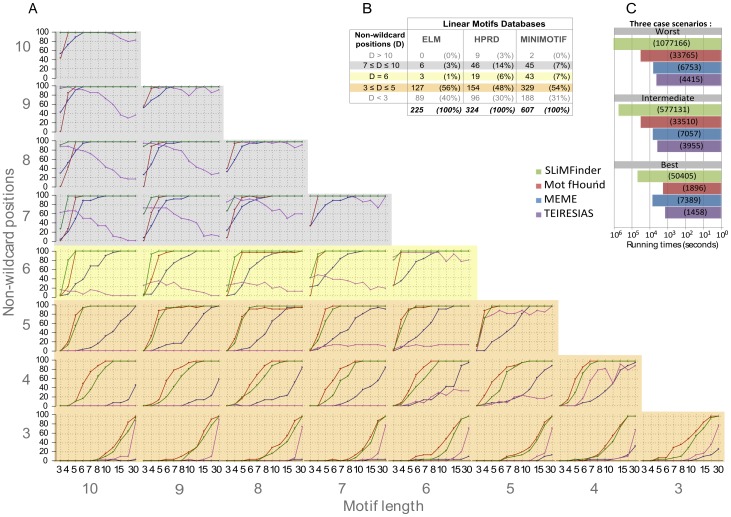
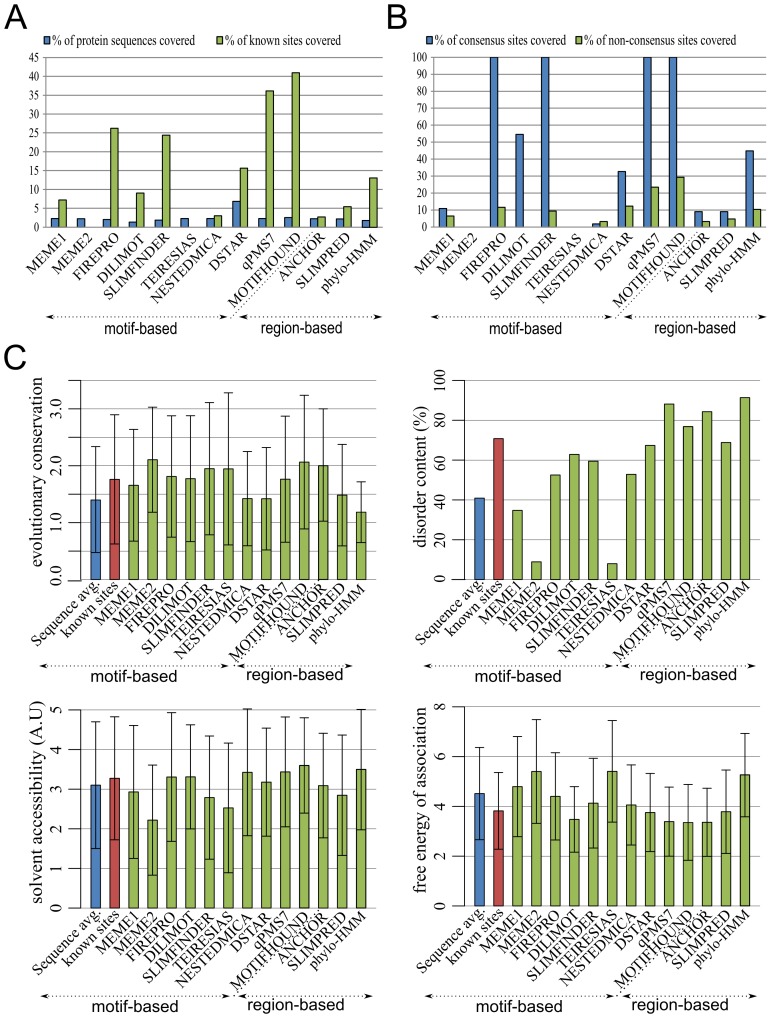
Linear motifs mediate a wide variety of cellular functions, which makes their characterization in protein sequences crucial to understanding cellular systems. However, the short length and degenerate nature of linear motifs make their discovery a difficult problem. Here, we introduce MotifHound, an algorithm particularly suited for the discovery of small and degenerate linear motifs. MotifHound performs an exact and exhaustive enumeration of all motifs present in proteins of interest, including all of their degenerate forms, and scores the overrepresentation of each motif based on its occurrence in proteins of interest relative to a background (e.g., proteome) using the hypergeometric distribution. To assess MotifHound, we benchmarked it together with state-of-the-art algorithms. The benchmark consists of 11,880 sets of proteins from S. cerevisiae; in each set, we artificially spiked-in one motif varying in terms of three key parameters, (i) number of occurrences, (ii) length and (iii) the number of degenerate or "wildcard" positions. The benchmark enabled the evaluation of the impact of these three properties on the performance of the different algorithms. The results showed that MotifHound and SLiMFinder were the most accurate in detecting degenerate linear motifs. Interestingly, MotifHound was 15 to 20 times faster at comparable accuracy and performed best in the discovery of highly degenerate motifs. We complemented the benchmark by an analysis of proteins experimentally shown to bind the FUS1 SH3 domain from S. cerevisiae. Using the full-length protein partners as sole information, MotifHound recapitulated most experimentally determined motifs binding to the FUS1 SH3 domain. Moreover, these motifs exhibited properties typical of SH3 binding peptides, e.g., high intrinsic disorder and evolutionary conservation, despite the fact that none of these properties were used as prior information. MotifHound is available (http://michnick.bcm.umontreal.ca or http://tinyurl.com/motifhound) together with the benchmark that can be used as a reference to assess future developments in motif discovery.
Conflict of interest statement
Figures
Similar articles
-
Exhaustive search of linear information encoding protein-peptide recognition.PLoS Comput Biol. 2017 Apr 20;13(4):e1005499. doi: 10.1371/journal.pcbi.1005499. eCollection 2017 Apr. PLoS Comput Biol. 2017. PMID: 28426660 Free PMC article.
-
SLiMFinder: a probabilistic method for identifying over-represented, convergently evolved, short linear motifs in proteins.PLoS One. 2007 Oct 3;2(10):e967. doi: 10.1371/journal.pone.0000967. PLoS One. 2007. PMID: 17912346 Free PMC article.
-
Bioinformatics Approaches for Predicting Disordered Protein Motifs.Adv Exp Med Biol. 2015;870:291-318. doi: 10.1007/978-3-319-20164-1_9. Adv Exp Med Biol. 2015. PMID: 26387106 Review.
-
Estimation and efficient computation of the true probability of recurrence of short linear protein sequence motifs in unrelated proteins.BMC Bioinformatics. 2010 Jan 7;11:14. doi: 10.1186/1471-2105-11-14. BMC Bioinformatics. 2010. PMID: 20055997 Free PMC article.
-
Computational identification and analysis of protein short linear motifs.Front Biosci (Landmark Ed). 2010 Jun 1;15(3):801-25. doi: 10.2741/3647. Front Biosci (Landmark Ed). 2010. PMID: 20515727 Review.
Cited by
-
FaSTPACE: a fast and scalable tool for peptide alignment and consensus extraction.NAR Genom Bioinform. 2024 Aug 21;6(3):lqae103. doi: 10.1093/nargab/lqae103. eCollection 2024 Sep. NAR Genom Bioinform. 2024. PMID: 39170861 Free PMC article.
-
High-throughput methods for identification of protein-protein interactions involving short linear motifs.Cell Commun Signal. 2015 Aug 22;13:38. doi: 10.1186/s12964-015-0116-8. Cell Commun Signal. 2015. PMID: 26297553 Free PMC article.
-
Probabilistic variable-length segmentation of protein sequences for discriminative motif discovery (DiMotif) and sequence embedding (ProtVecX).Sci Rep. 2019 Mar 5;9(1):3577. doi: 10.1038/s41598-019-38746-w. Sci Rep. 2019. PMID: 30837494 Free PMC article.
-
HH-MOTiF: de novo detection of short linear motifs in proteins by Hidden Markov Model comparisons.Nucleic Acids Res. 2017 Jul 3;45(W1):W470-W477. doi: 10.1093/nar/gkx341. Nucleic Acids Res. 2017. PMID: 28460141 Free PMC article.
-
A bioinformatics pipeline to search functional motifs within whole-proteome data: a case study of poxviruses.Virus Genes. 2017 Apr;53(2):173-178. doi: 10.1007/s11262-016-1416-9. Epub 2016 Dec 20. Virus Genes. 2017. PMID: 28000080 Free PMC article.
References
-
- Diella F, Haslam N, Chica C, Budd A, Michael S, et al. (2008) Understanding eukaryotic linear motifs and their role in cell signaling and regulation. Front Biosci 13: 6580–6603. - PubMed
-
- Davey NE, Van Roey K, Weatheritt RJ, Toedt G, Uyar B, et al. (2012) Attributes of short linear motifs. Mol Biosyst 8: 268–281. - PubMed
-
- Davey NE, Edwards RJ, Shields DC (2010) Computational identification and analysis of protein short linear motifs. Front Biosci 15: 801–825. - PubMed
-
- Van Roey K, Gibson TJ, Davey NE (2012) Motif switches: decision-making in cell regulation. Curr Opin Struct Biol 22: 378–385. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
