

Wolfram syndrome in the Japanese population; molecular analysis of WFS1 gene and characterization of clinical features
- PMID: 25211237
- PMCID: PMC4161373
- DOI: 10.1371/journal.pone.0106906
Wolfram syndrome in the Japanese population; molecular analysis of WFS1 gene and characterization of clinical features
Abstract
Background: Wolfram syndrome (WFS) is a recessive neurologic and endocrinologic degenerative disorder, and is also known as DIDMOAD (Diabetes Insipidus, early-onset Diabetes Mellitus, progressive Optic Atrophy and Deafness) syndrome. Most affected individuals carry recessive mutations in the Wolfram syndrome 1 gene (WFS1). However, the phenotypic pleiomorphism, rarity and molecular complexity of this disease complicate our efforts to understand WFS. To address this limitation, we aimed to describe complications and to elucidate the contributions of WFS1 mutations to clinical manifestations in Japanese patients with WFS.
Methodology: The minimal ascertainment criterion for diagnosing WFS was having both early onset diabetes mellitus and bilateral optic atrophy. Genetic analysis for WFS1 was performed by direct sequencing.
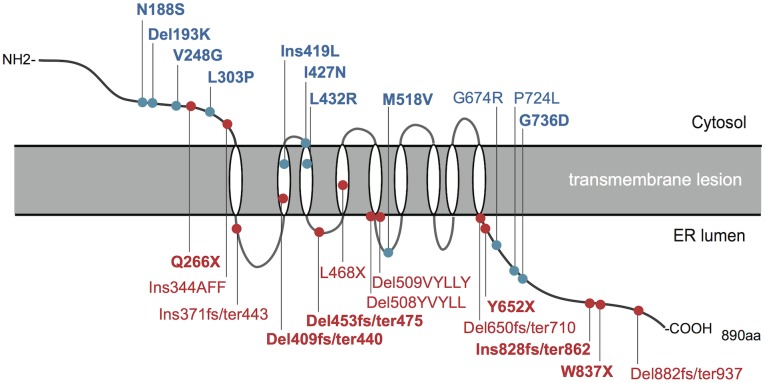
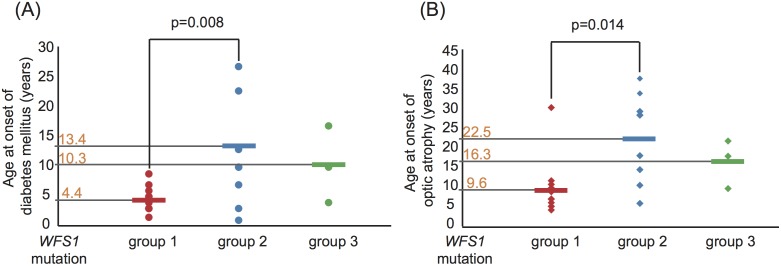
Principal findings: Sixty-seven patients were identified nationally for a prevalence of one per 710,000, with 33 patients (49%) having all 4 components of DIDMOAD. In 40 subjects who agreed to participate in this investigation from 30 unrelated families, the earliest manifestation was DM at a median age of 8.7 years, followed by OA at a median age of 15.8 years. However, either OA or DI was the first diagnosed feature in 6 subjects. In 10, features other than DM predated OA. Twenty-seven patients (67.5%) had a broad spectrum of recessive mutations in WFS1. Two patients had mutations in only one allele. Eleven patients (27.5%) had intact WFS1 alleles. Ages at onset of both DM and OA in patients with recessive WFS1 mutations were indistinguishable from those in patients without WFS1 mutations. In the patients with predicted complete loss-of-function mutations, ages at the onsets of both DM and OA were significantly earlier than those in patients with predicted partial-loss-of function mutations.
Conclusion/significance: This study emphasizes the clinical and genetic heterogeneity in patients with WFS. Genotype-phenotype correlations may exist in patients with WFS1 mutations, as demonstrated by the disease onset.
Conflict of interest statement
Figures
References
-
- Wolfram DJ, Wagener H (1938) Diabetes mellitus and simple optic atrophy among siblings. A report of four cases. Proc Mayo Clin 13: 715–718.
-
- Barrett TG, Bundey SE, Macleod AF (1995) Neurodegeneration and diabetes: UK nationwide study of Wolfram (DIDMOAD) syndrome. Lancet 346: 1458–1463. - PubMed
-
- Minton JA, Rainbow LA, Ricketts C, Barrett TG (2003) Wolfram syndrome. Rev Endocr Metab Disord 4: 53–59. - PubMed
-
- de Heredia ML, Cleries R, Nunes V (2013) Genotypic classification of patients with Wolfram syndrome: insights into the natural history of the disease and correlation with phenotype. Genet Med 15: 497–506. - PubMed
-
- Inoue H, Tanizawa Y, Wasson J, Behn P, Kalidas K, et al. (1998) A gene encoding a transmembrane protein is mutated in patients with diabetes mellitus and optic atrophy (Wolfram syndrome). Nat Genet 20: 143–148. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
