

Obesity, insulin resistance and comorbidities? Mechanisms of association
- PMID: 25211442
- PMCID: PMC4423826
- DOI: 10.1590/0004-2730000003223
Obesity, insulin resistance and comorbidities? Mechanisms of association
Abstract
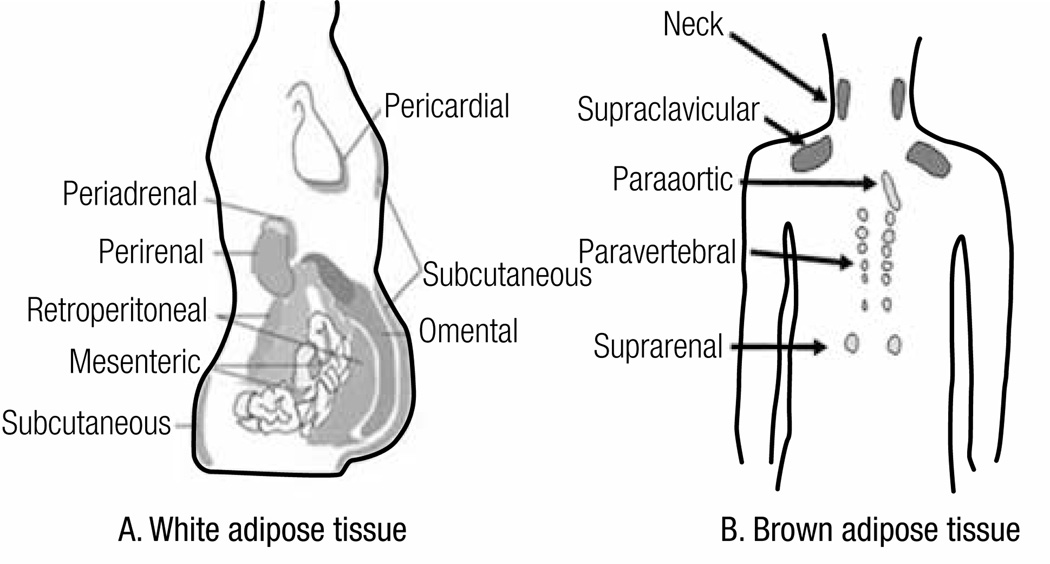
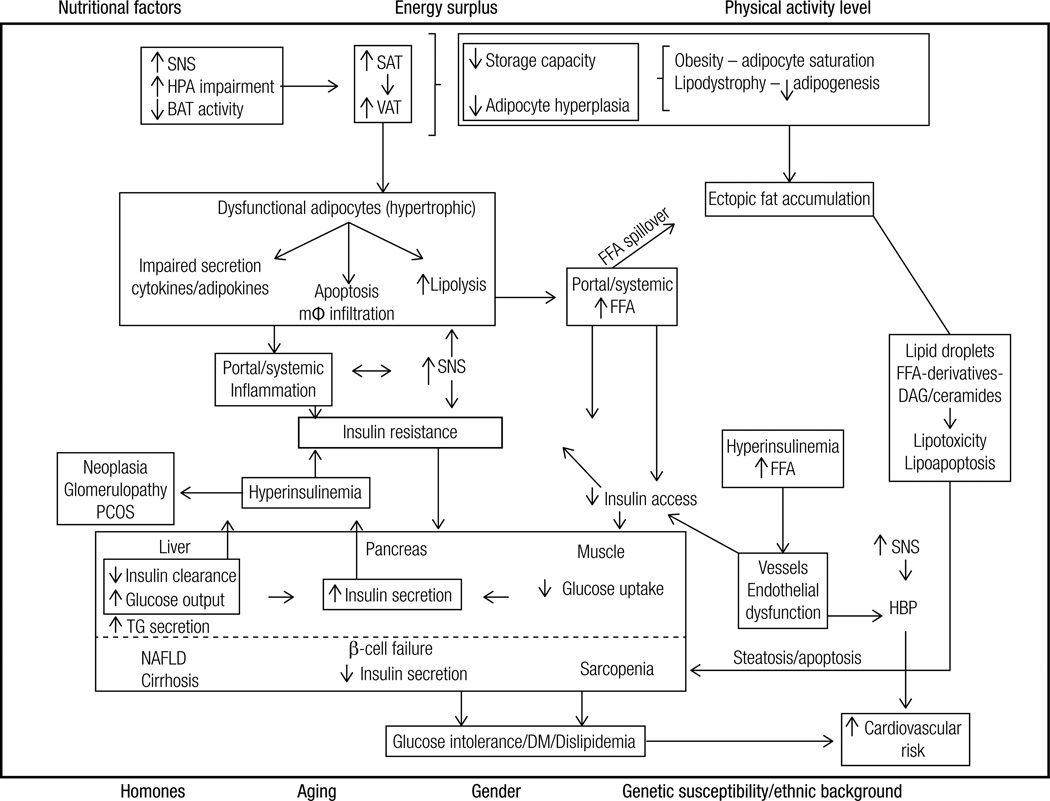
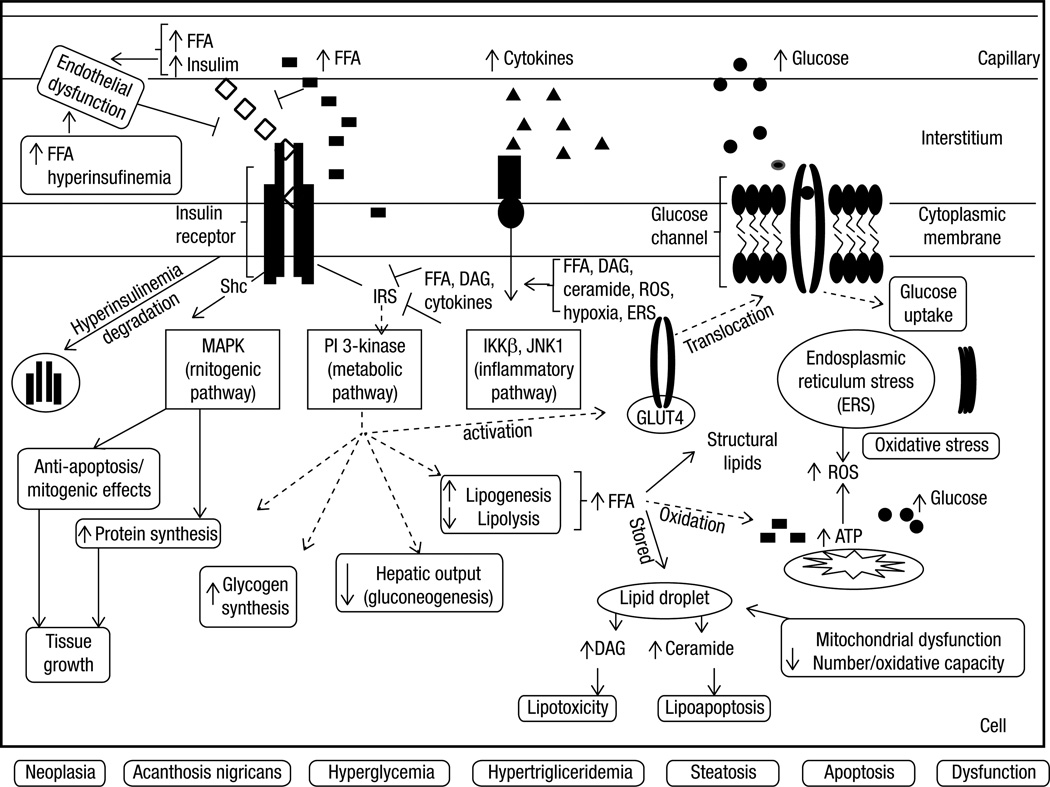
Overall excess of fat, usually defined by the body mass index, is associated with metabolic (e.g. glucose intolerance, type 2 diabetes mellitus (T2DM), dyslipidemia) and non-metabolic disorders (e.g. neoplasias, polycystic ovary syndrome, non-alcoholic fat liver disease, glomerulopathy, bone fragility etc.). However, more than its total amount, the distribution of adipose tissue throughout the body is a better predictor of the risk to the development of those disorders. Fat accumulation in the abdominal area and in non-adipose tissue (ectopic fat), for example, is associated with increased risk to develop metabolic and non-metabolic derangements. On the other hand, observations suggest that individuals who present peripheral adiposity, characterized by large hip and thigh circumferences, have better glucose tolerance, reduced incidence of T2DM and of metabolic syndrome. Insulin resistance (IR) is one of the main culprits in the association between obesity, particularly visceral, and metabolic as well as non-metabolic diseases. In this review we will highlight the current pathophysiological and molecular mechanisms possibly involved in the link between increased VAT, ectopic fat, IR and comorbidities. We will also provide some insights in the identification of these abnormalities.
Conflict of interest statement
Disclosure: no potential conflict of interest relevant to this article was reported.
Figures
References
-
- Tchernof A, Despres JP. Pathophysiology of human visceral obesity: an update. Physiol Rev. 2013;93(1):359–404. - PubMed
-
- Vazquez G, Duval S, Jacobs DR, Jr, Silventoinen K. Comparison of body mass index, waist circumference, and waist/hip ratio in predicting incident diabetes: a meta-analysis. Epidemiol Rev. 2007;29:115–128. - PubMed
-
- Després JP. Body fat distribution and risk of cardiovascular disease: an update. Circulation. 2012;126(10):1301–1313. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical