

Elevation of brain magnesium prevents synaptic loss and reverses cognitive deficits in Alzheimer's disease mouse model
- PMID: 25213836
- PMCID: PMC4172865
- DOI: 10.1186/s13041-014-0065-y
Elevation of brain magnesium prevents synaptic loss and reverses cognitive deficits in Alzheimer's disease mouse model
Abstract
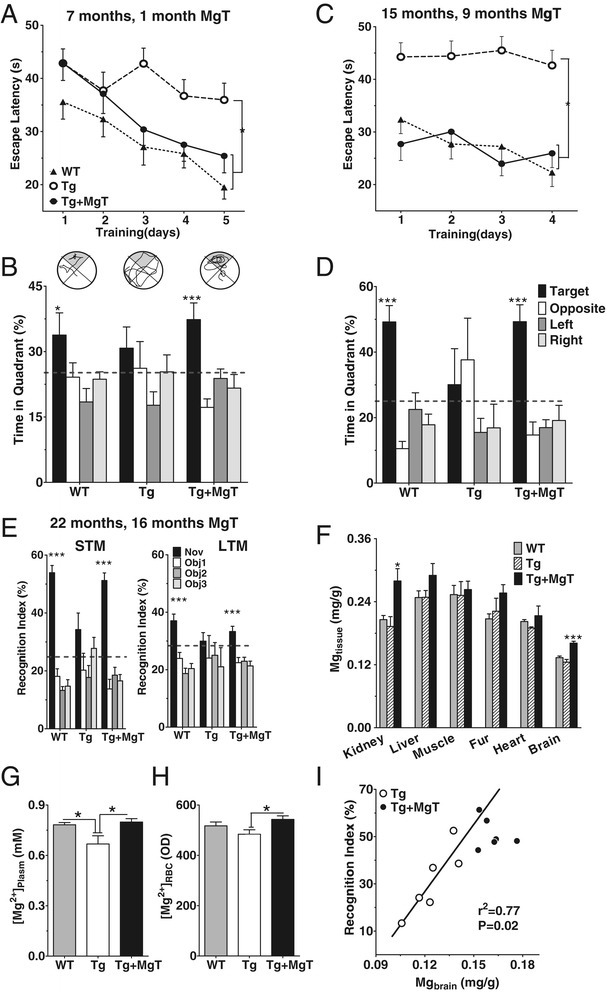
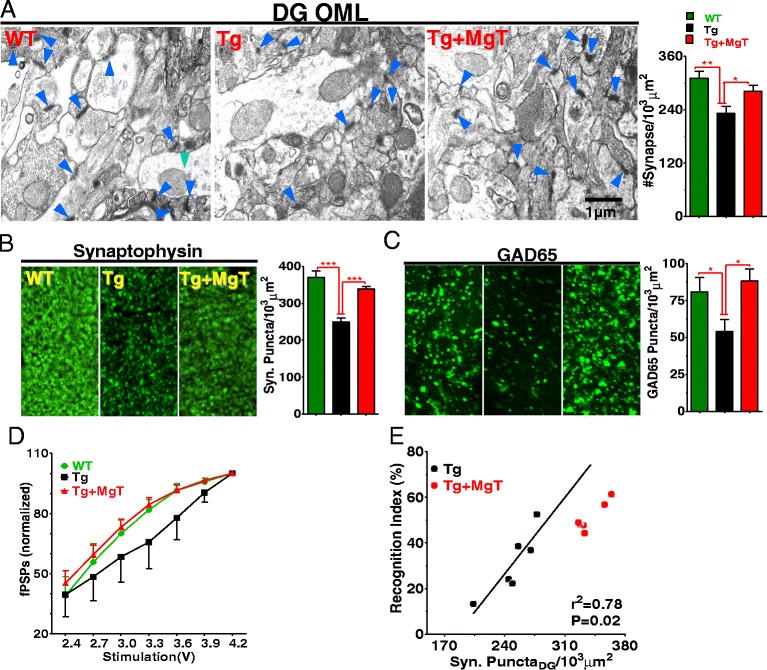
Background: Profound synapse loss is one of the major pathological hallmarks associated with Alzheimer's disease, which might underlie memory impairment. Our previous work demonstrates that magnesium ion is a critical factor in controlling synapse density/plasticity. Here, we tested whether elevation of brain magnesium, using a recently developed compound (magnesium-L-threonate, MgT), can ameliorate the AD-like pathologies and cognitive deficits in the APPswe/PS1dE9 mice, a transgenic mouse model of Alzheimer's disease.
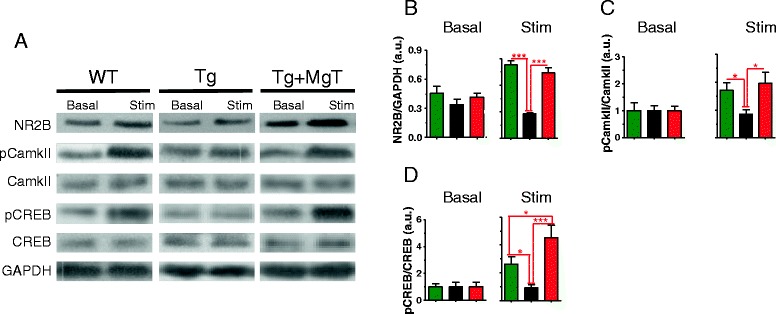
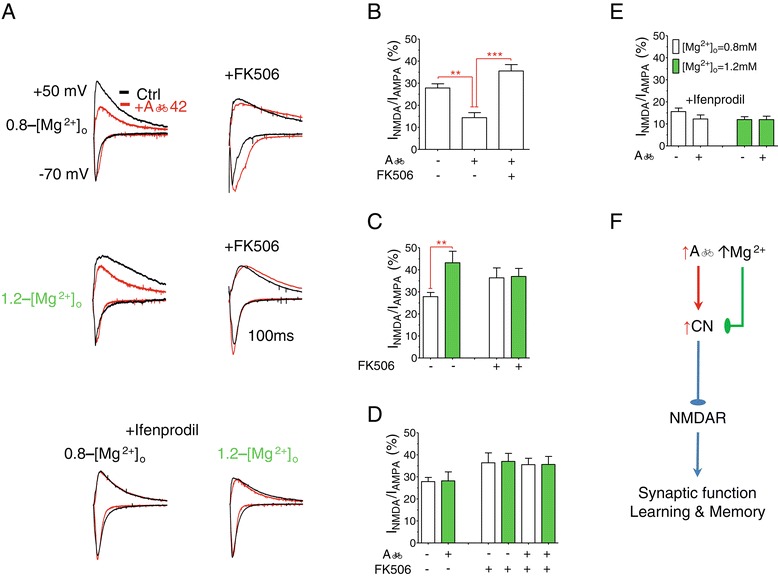
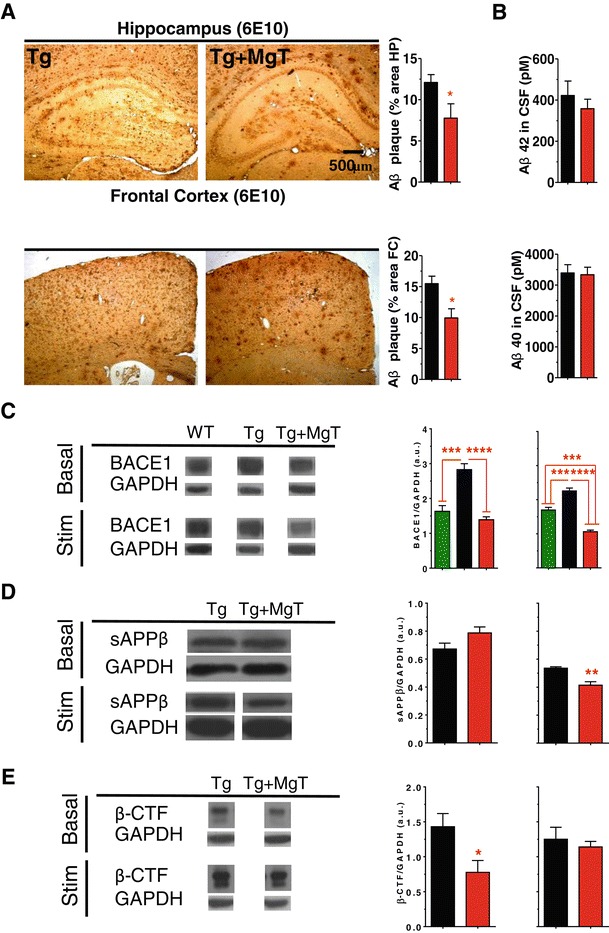
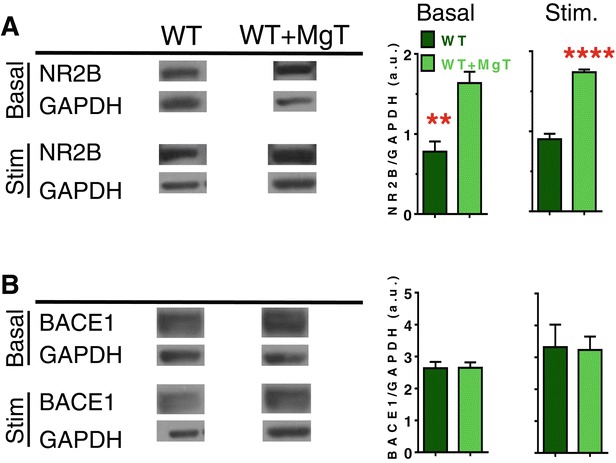
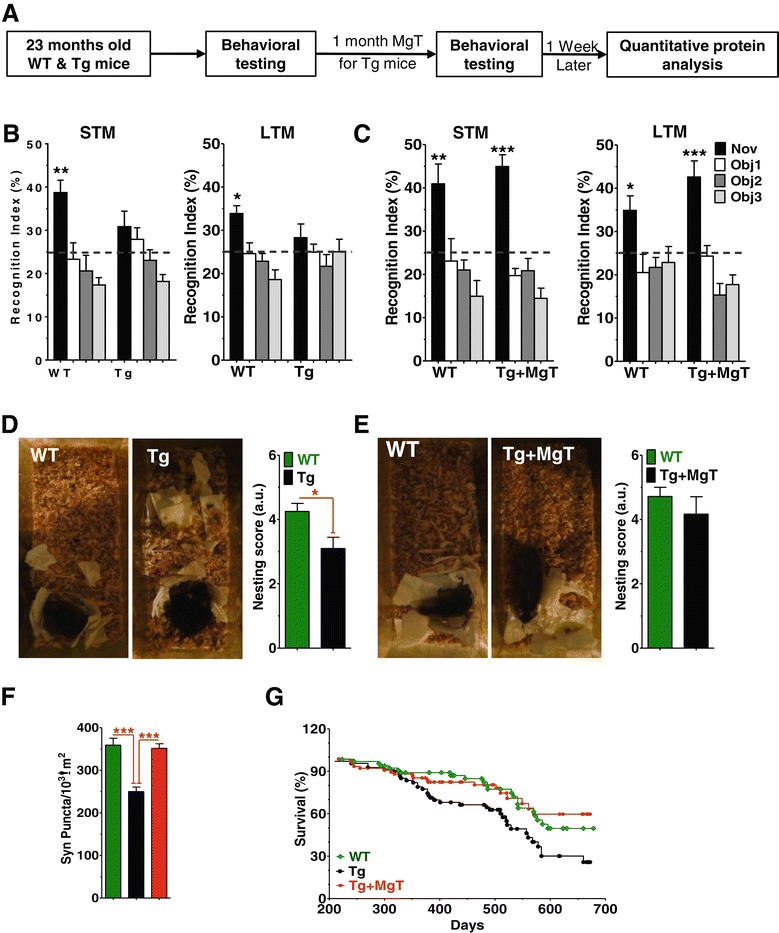
Results: MgT treatment reduced Aβ-plaque, prevented synapse loss and memory decline in the transgenic mice. Strikingly, MgT treatment was effective even when the treatment was given to the mice at the end-stage of their Alzheimer's disease-like pathological progression. To explore how elevation of brain magnesium ameliorates the AD-like pathologies in the brain of transgenic mice, we studied molecules critical for APP metabolism and signaling pathways implicated in synaptic plasticity/density. In the transgenic mice, the NMDAR signaling pathway was downregulated, while the BACE1 expression were upregulated. MgT treatment prevented the impairment of these signaling pathways, stabilized BACE1 expression and reduced sAPPβ and β-CTF in the transgenic mice. At the molecular level, elevation of extracellular magnesium prevented the high Aβ-induced reductions in synaptic NMDARs by preventing calcineurin overactivation in hippocampal slices.
Conclusions: Our results suggest that elevation of brain magnesium exerts substantial synaptoprotective effects in a mouse model of Alzheimer's disease, and hence it might have therapeutic potential for treating Alzheimer's disease.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
