

Tissue-specific responses to the LRPPRC founder mutation in French Canadian Leigh Syndrome
- PMID: 25214534
- PMCID: PMC4275074
- DOI: 10.1093/hmg/ddu468
Tissue-specific responses to the LRPPRC founder mutation in French Canadian Leigh Syndrome
Abstract
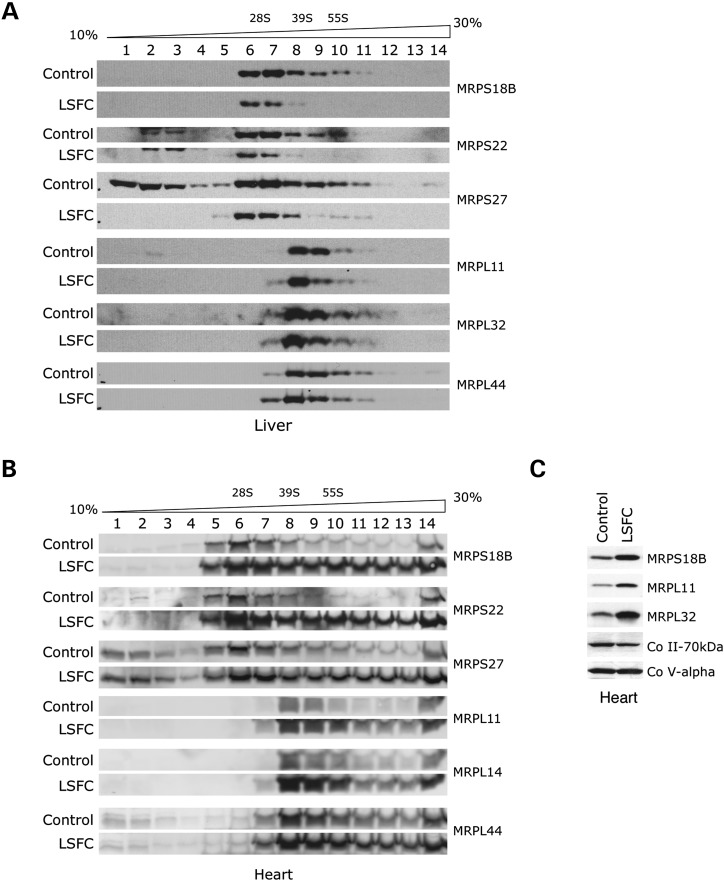
French Canadian Leigh Syndrome (LSFC) is an early-onset, progressive neurodegenerative disorder with a distinct pattern of tissue involvement. Most cases are caused by a founder missense mutation in LRPPRC. LRPPRC forms a ribonucleoprotein complex with SLIRP, another RNA-binding protein, and this stabilizes polyadenylated mitochondrial mRNAs. LSFC fibroblasts have reduced levels of LRPPRC and a specific complex IV assembly defect; however, further depletion of mutant LRPPRC results in a complete failure to assemble a functional oxidative phosphorylation system, suggesting that LRPPRC levels determine the nature of the biochemical phenotype. We tested this hypothesis in cultured muscle cells and tissues from LSFC patients. LRPPRC levels were reduced in LSFC muscle cells, resulting in combined complex I and IV deficiencies. A similar combined deficiency was observed in skeletal muscle. Complex IV was only moderately reduced in LSFC heart, but was almost undetectable in liver. Both of these tissues showed elevated levels of complexes I and III. Despite the marked biochemical differences, the steady-state levels of LRPPRC and mitochondrial mRNAs were extremely low, LRPPRC was largely detergent-insoluble, and SLIRP was undetectable in all LSFC tissues. The level of the LRPPRC/SLIRP complex appeared much reduced in control tissues by the first dimension blue-native polyacrylamide gel electrophoresis (BN-PAGE) analysis compared with fibroblasts, and even by second dimension analysis it was virtually undetectable in control heart. These results point to tissue-specific pathways for the post-transcriptional handling of mitochondrial mRNAs and suggest that the biochemical defects in LSFC reflect the differential ability of tissues to adapt to the mutation.
© The Author 2014. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures








References
-
- Koopman W.J., Willems P.H., Smeitink J.A. Monogenic mitochondrial disorders. N. Engl. J. Med. 2012;366:1132–1141. - PubMed
-
- Zhu Z., Yao J., Johns T., Fu K., De Bie I., Macmillan C., Cuthbert A.P., Newbold R.F., Wang J., Chevrette M., et al. SURF1, encoding a factor involved in the biogenesis of cytochrome c oxidase, is mutated in Leigh syndrome. Nat. Genet. 1998;20:337–343. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
