

Loss of nNOS inhibits compensatory muscle hypertrophy and exacerbates inflammation and eccentric contraction-induced damage in mdx mice
- PMID: 25214536
- PMCID: PMC4275075
- DOI: 10.1093/hmg/ddu469
Loss of nNOS inhibits compensatory muscle hypertrophy and exacerbates inflammation and eccentric contraction-induced damage in mdx mice
Abstract
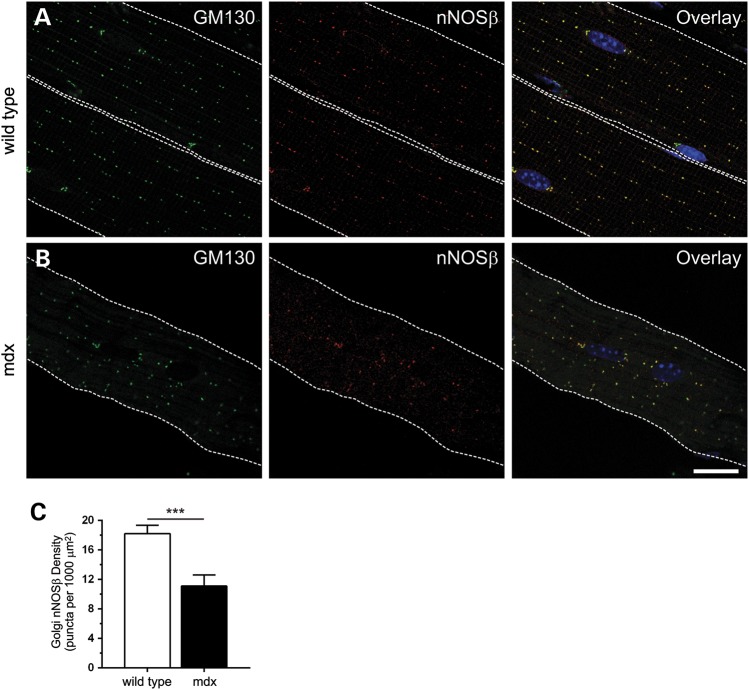
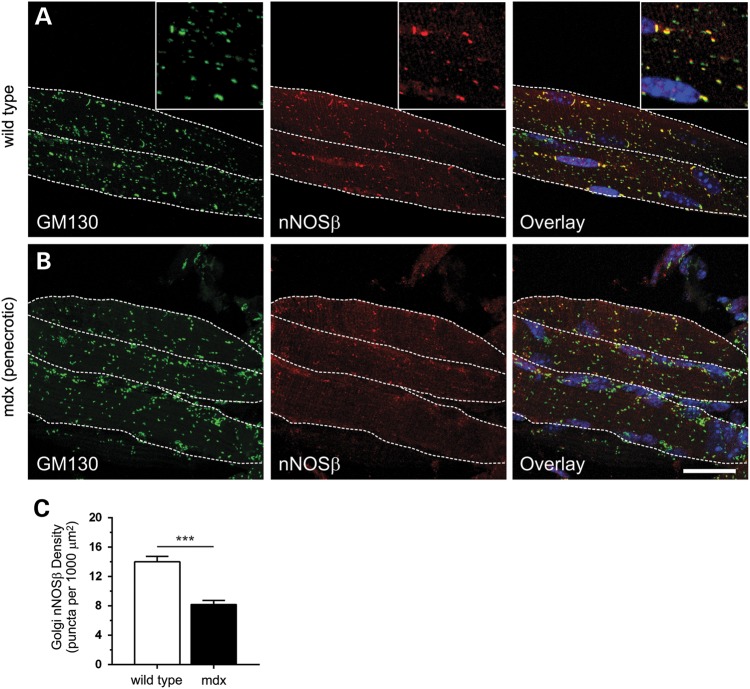
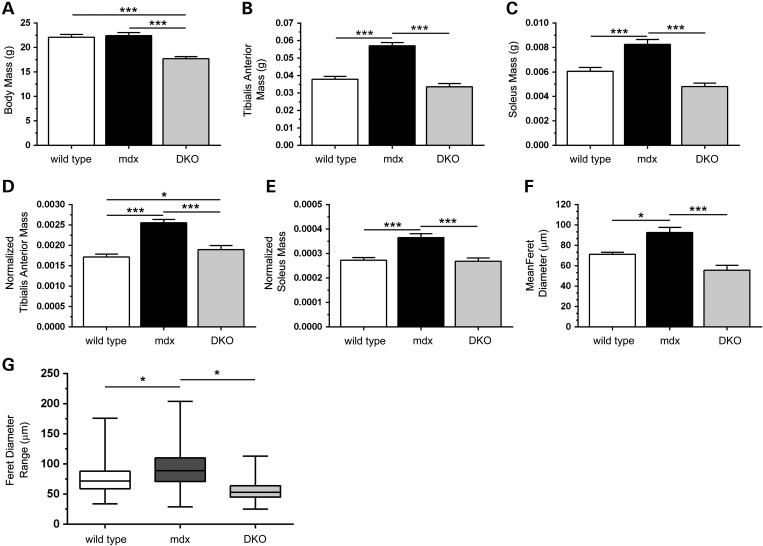
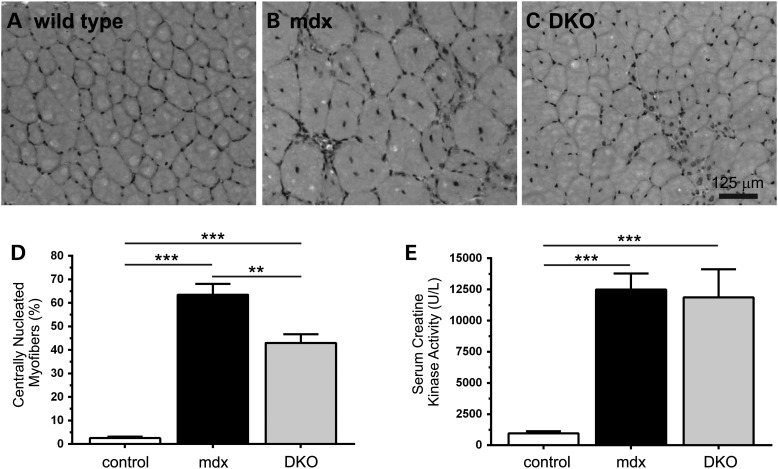
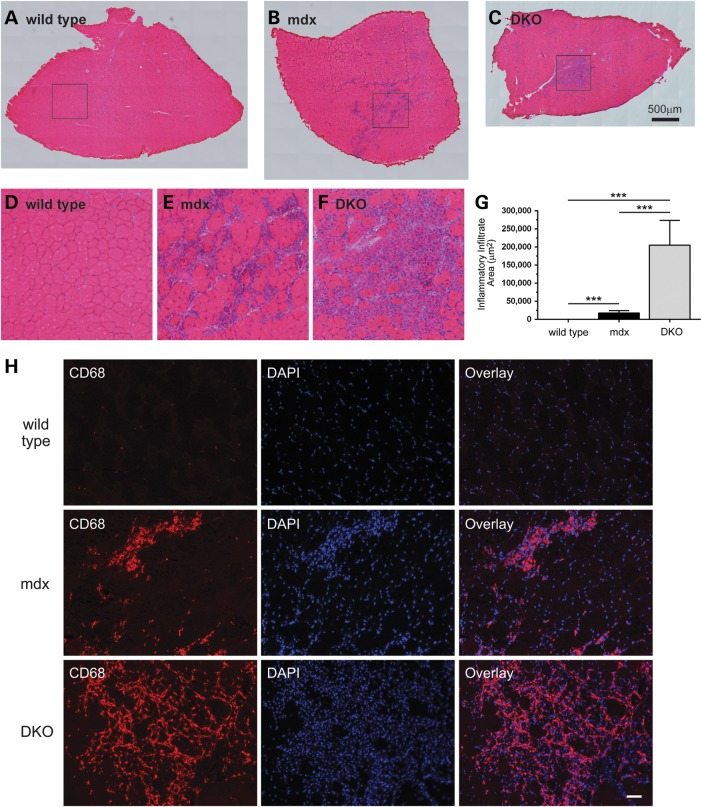
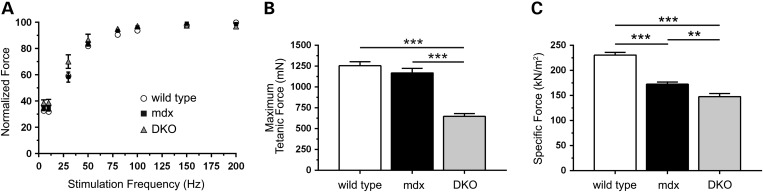
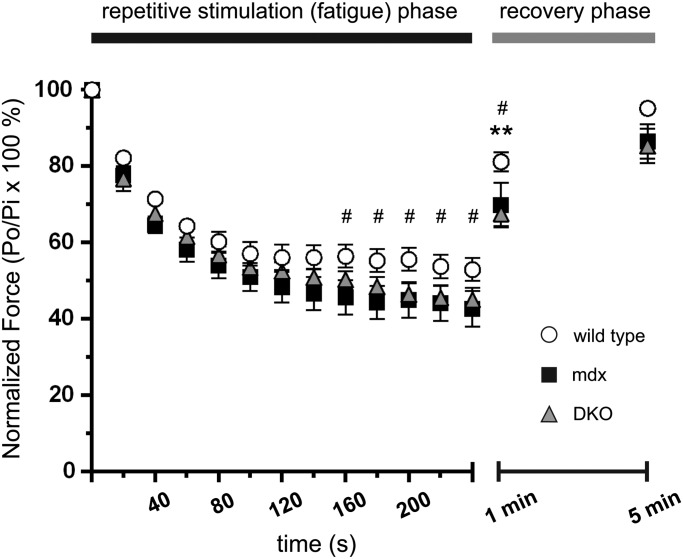
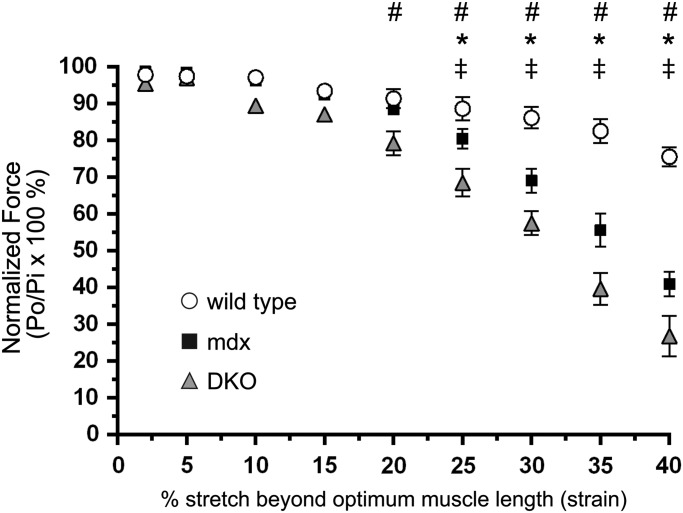
Approaches targeting nitric oxide (NO) signaling show promise as therapies for Duchenne and Becker muscular dystrophies. However, the mechanisms by which NO benefits dystrophin-deficient muscle remain unclear, but may involve nNOSβ, a newly discovered enzymatic source of NO in skeletal muscle. Here we investigate the impact of dystrophin deficiency on nNOSβ and use mdx mice engineered to lack nNOSμ and nNOSβ to discern how the loss of nNOS impacts dystrophic skeletal muscle pathology. In mdx muscle, nNOSβ was mislocalized and its association with the Golgi complex was reduced. nNOS depletion from mdx mice prevented compensatory skeletal muscle cell hypertrophy, decreased myofiber central nucleation and increased focal macrophage cell infiltration, indicating exacerbated dystrophic muscle damage. Reductions in muscle integrity in nNOS-null mdx mice were accompanied by decreases in specific force and increased susceptibility to eccentric contraction-induced muscle damage compared with mdx controls. Unexpectedly, muscle fatigue was unaffected by nNOS depletion, revealing a novel latent compensatory mechanism for the loss of nNOS in mdx mice. Together with previous studies, these data suggest that localization of both nNOSμ and nNOSβ is disrupted by dystrophin deficiency. They also indicate that nNOS has a more complex role as a modifier of dystrophic pathology and broader therapeutic potential than previously recognized. Importantly, these findings also suggest nNOSβ as a new drug target and provide a new conceptual framework for understanding nNOS signaling and the benefits of NO therapies in dystrophinopathies.
© The Author 2014. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures
Similar articles
-
A nitric oxide synthase transgene ameliorates muscular dystrophy in mdx mice.J Cell Biol. 2001 Oct 1;155(1):123-31. doi: 10.1083/jcb.200105110. J Cell Biol. 2001. PMID: 11581289 Free PMC article.
-
Dual AAV therapy ameliorates exercise-induced muscle injury and functional ischemia in murine models of Duchenne muscular dystrophy.Hum Mol Genet. 2013 Sep 15;22(18):3720-9. doi: 10.1093/hmg/ddt224. Epub 2013 May 15. Hum Mol Genet. 2013. PMID: 23681067 Free PMC article.
-
Xanthine oxidase is hyper-active in Duchenne muscular dystrophy.Free Radic Biol Med. 2018 Dec;129:364-371. doi: 10.1016/j.freeradbiomed.2018.10.404. Epub 2018 Oct 10. Free Radic Biol Med. 2018. PMID: 30312761 Free PMC article.
-
Neuronal nitric oxide synthase (nNOS) splice variant function: Insights into nitric oxide signaling from skeletal muscle.Nitric Oxide. 2019 Jan 1;82:35-47. doi: 10.1016/j.niox.2018.11.004. Epub 2018 Nov 29. Nitric Oxide. 2019. PMID: 30503614 Review.
-
Nitric oxide synthase deficiency and the pathophysiology of muscular dystrophy.J Physiol. 2014 Nov 1;592(21):4627-38. doi: 10.1113/jphysiol.2014.274878. Epub 2014 Sep 5. J Physiol. 2014. PMID: 25194047 Free PMC article. Review.
Cited by
-
Nitric Oxide Regulates Skeletal Muscle Fatigue, Fiber Type, Microtubule Organization, and Mitochondrial ATP Synthesis Efficiency Through cGMP-Dependent Mechanisms.Antioxid Redox Signal. 2017 Jun 10;26(17):966-985. doi: 10.1089/ars.2016.6630. Epub 2016 Aug 17. Antioxid Redox Signal. 2017. PMID: 27393340 Free PMC article. Review.
-
Enhanced dimethylarginine degradation improves coronary flow reserve and exercise tolerance in Duchenne muscular dystrophy carrier mice.Am J Physiol Heart Circ Physiol. 2020 Sep 1;319(3):H582-H603. doi: 10.1152/ajpheart.00333.2019. Epub 2020 Aug 7. Am J Physiol Heart Circ Physiol. 2020. PMID: 32762558 Free PMC article.
-
Piezo1-mediated stellate cell activation causes pressure-induced pancreatic fibrosis in mice.JCI Insight. 2022 Apr 22;7(8):e158288. doi: 10.1172/jci.insight.158288. JCI Insight. 2022. PMID: 35451372 Free PMC article.
-
PM2.5 exposure induces vascular dysfunction via NO generated by iNOS in lung of ApoE-/- mouse.Int J Biol Sci. 2020 Jan 1;16(1):49-60. doi: 10.7150/ijbs.36073. eCollection 2020. Int J Biol Sci. 2020. PMID: 31892845 Free PMC article.
-
Nitric Oxide and Mechano-Electrical Transduction in Cardiomyocytes.Front Physiol. 2020 Dec 15;11:606740. doi: 10.3389/fphys.2020.606740. eCollection 2020. Front Physiol. 2020. PMID: 33384614 Free PMC article. Review.
References
-
- Koenig M., Hoffman E.P., Bertelson C.J., Monaco A.P., Feener C., Kunkel L.M. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987;50:509–517. - PubMed
-
- Hoffman E.P., Brown R.H., Jr, Kunkel L.M. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. - PubMed
-
- Brenman J.E., Chao D.S., Xia H., Aldape K., Bredt D.S. Nitric oxide synthase complexed with dystrophin and absent from skeletal muscle sarcolemma in Duchenne muscular dystrophy. Cell. 1995;82:743–752. - PubMed
-
- Crosbie R.H., Barresi R., Campbell K.P. Loss of sarcolemma nNOS in sarcoglycan-deficient muscle. FASEB J. 2002;16:1786–1791. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
