

Multilayered genetic and omics dissection of mitochondrial activity in a mouse reference population
- PMID: 25215496
- PMCID: PMC4179868
- DOI: 10.1016/j.cell.2014.07.039
Multilayered genetic and omics dissection of mitochondrial activity in a mouse reference population
Abstract
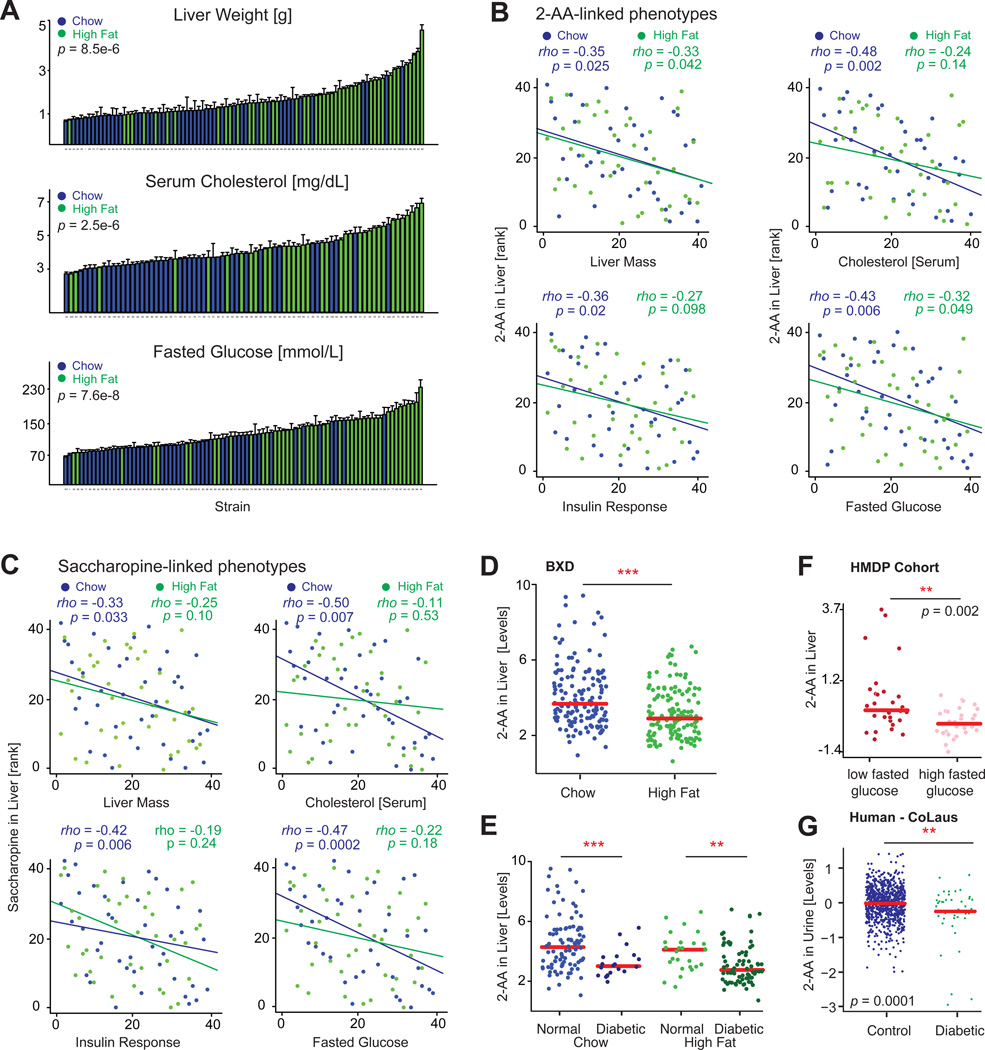
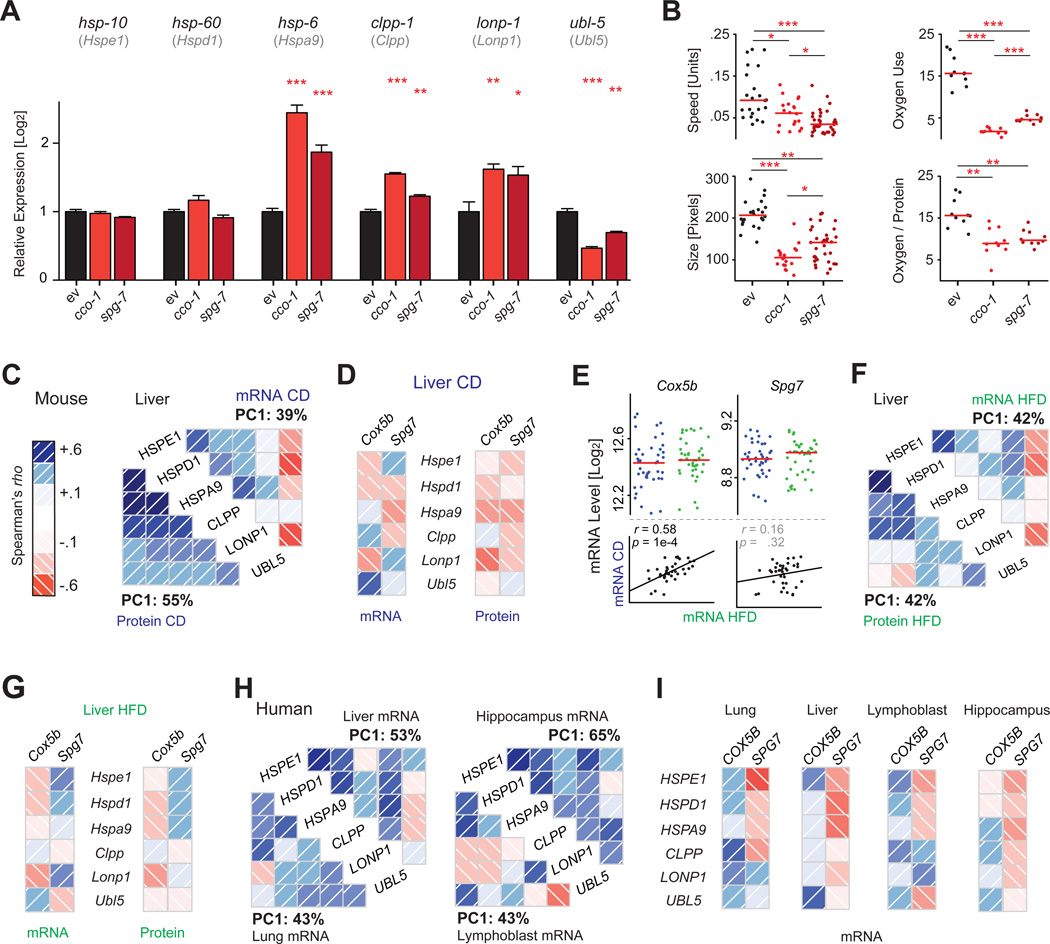
The manner by which genotype and environment affect complex phenotypes is one of the fundamental questions in biology. In this study, we quantified the transcriptome--a subset of the metabolome--and, using targeted proteomics, quantified a subset of the liver proteome from 40 strains of the BXD mouse genetic reference population on two diverse diets. We discovered dozens of transcript, protein, and metabolite QTLs, several of which linked to metabolic phenotypes. Most prominently, Dhtkd1 was identified as a primary regulator of 2-aminoadipate, explaining variance in fasted glucose and diabetes status in both mice and humans. These integrated molecular profiles also allowed further characterization of complex pathways, particularly the mitochondrial unfolded protein response (UPR(mt)). UPR(mt) shows strikingly variant responses at the transcript and protein level that are remarkably conserved among C. elegans, mice, and humans. Overall, these examples demonstrate the value of an integrated multilayered omics approach to characterize complex metabolic phenotypes.
Copyright © 2014 Elsevier Inc. All rights reserved.
Figures
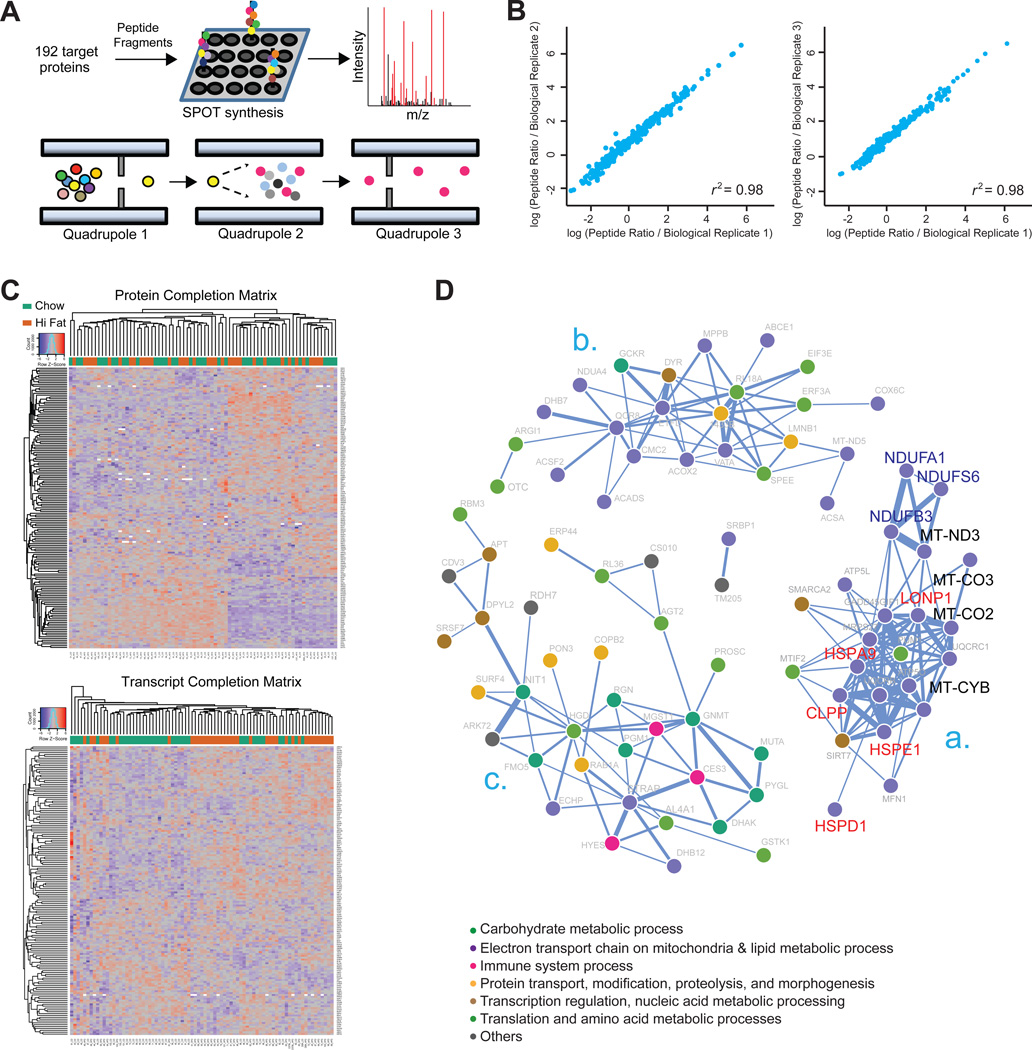
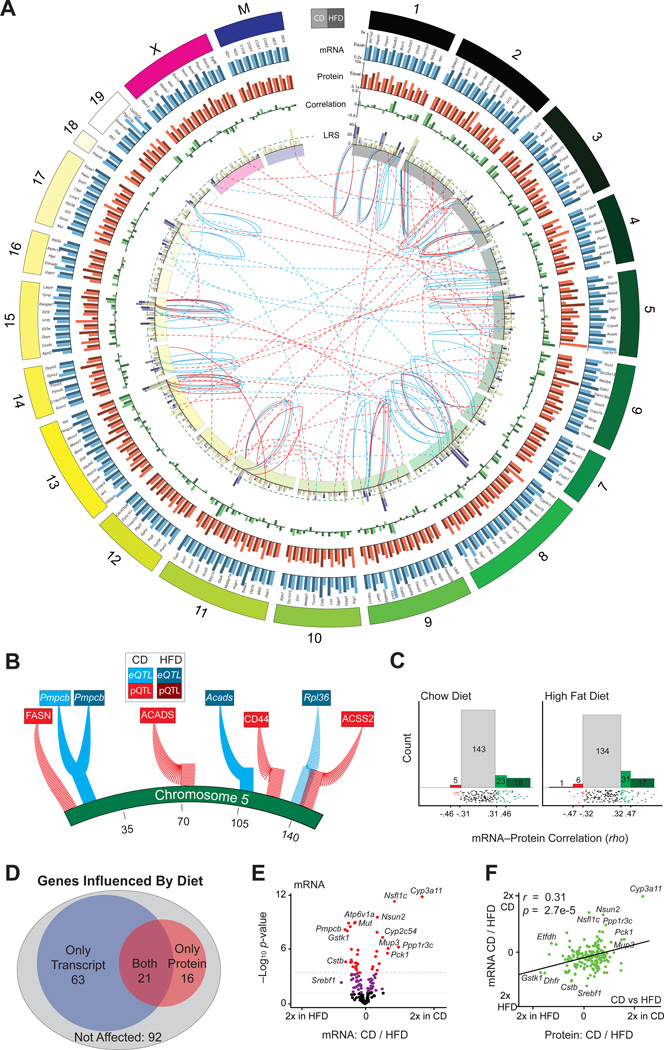
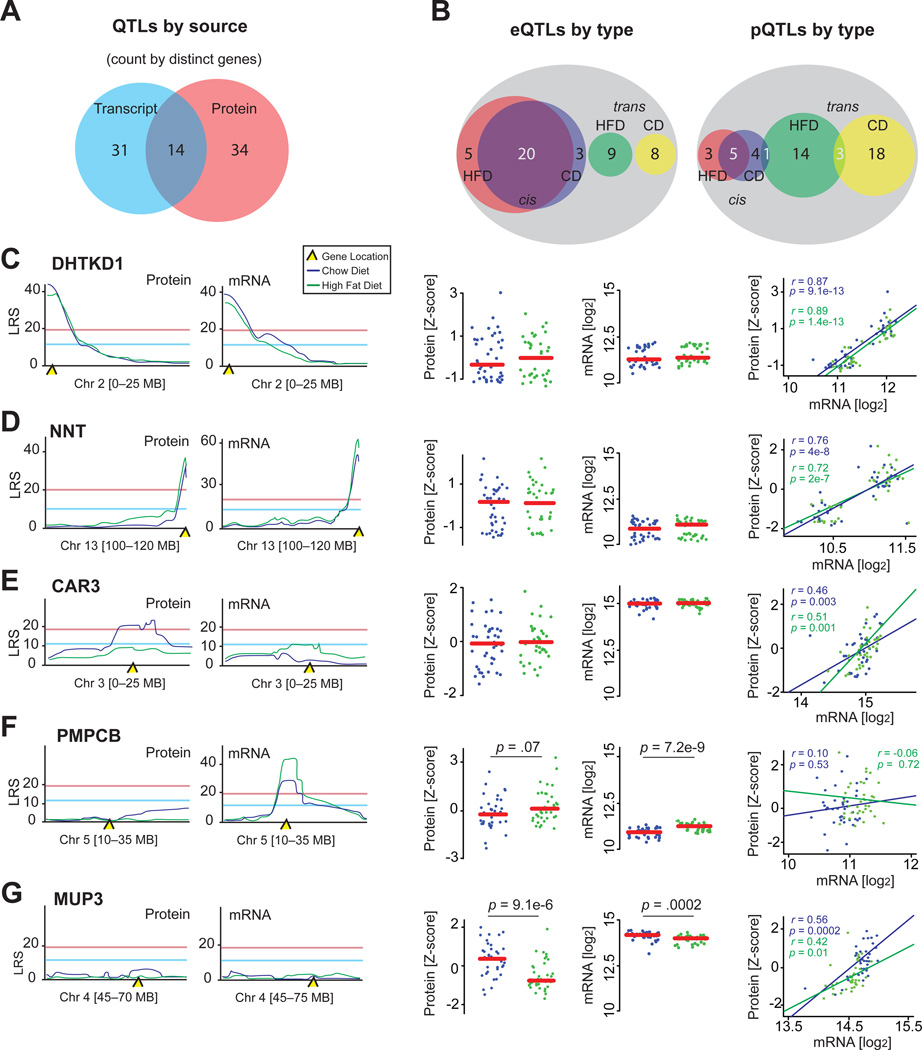
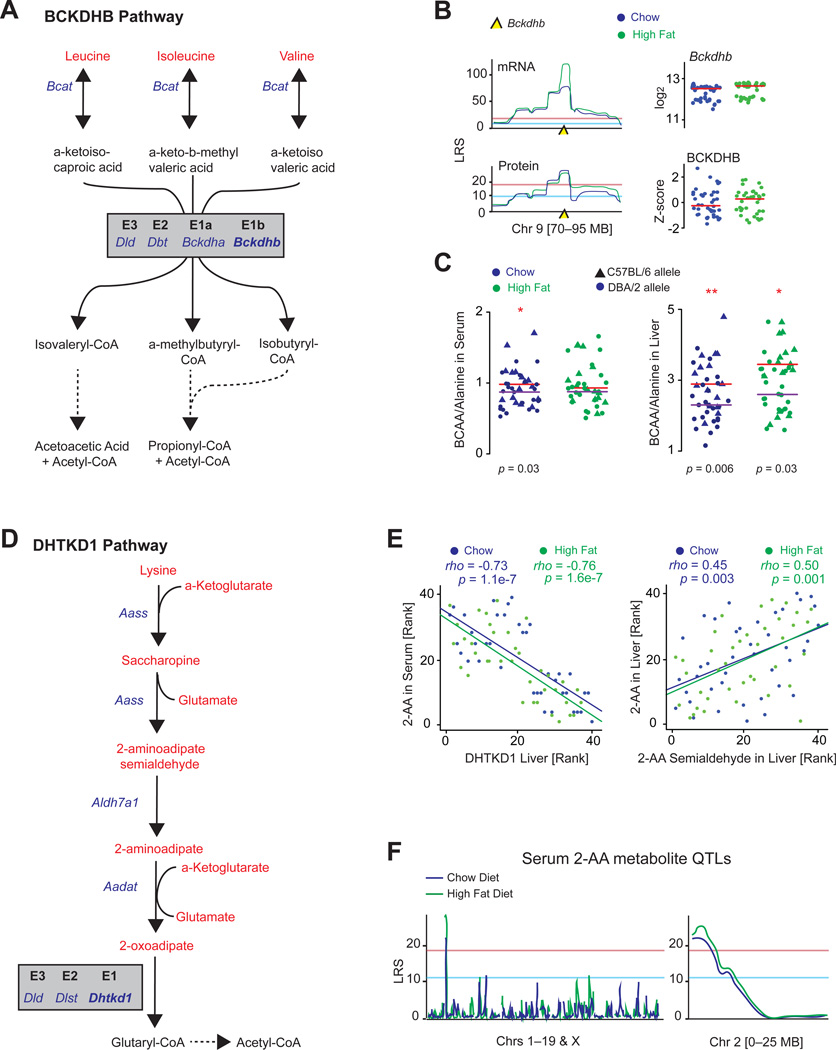
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
