

Determination and inference of eukaryotic transcription factor sequence specificity
- PMID: 25215497
- PMCID: PMC4163041
- DOI: 10.1016/j.cell.2014.08.009
Determination and inference of eukaryotic transcription factor sequence specificity
Abstract
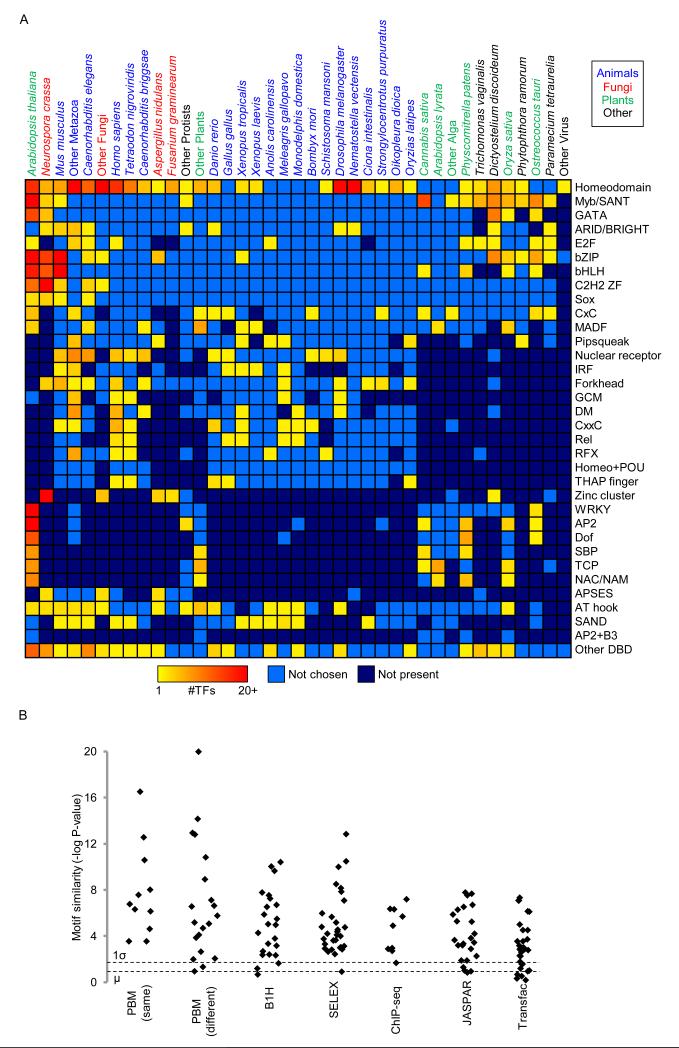
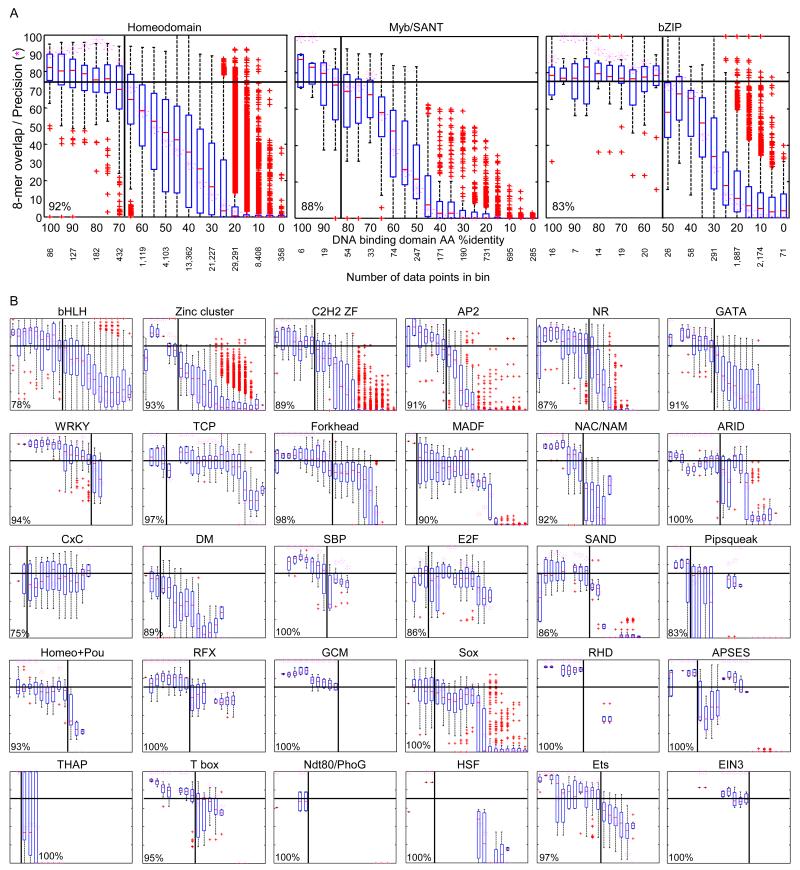
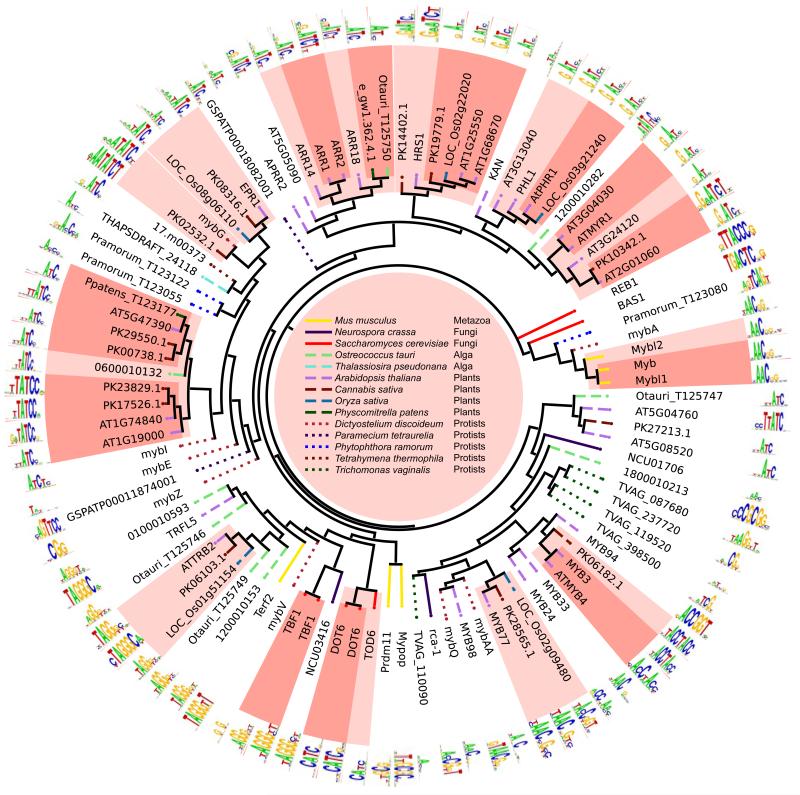
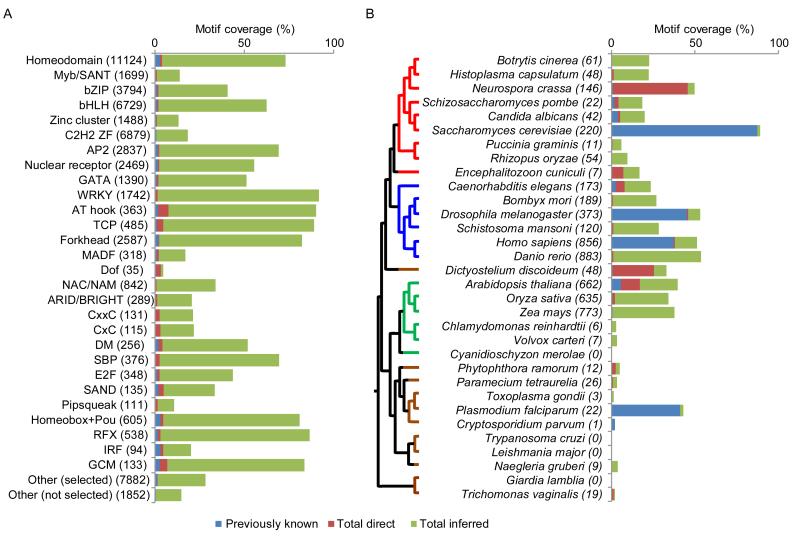
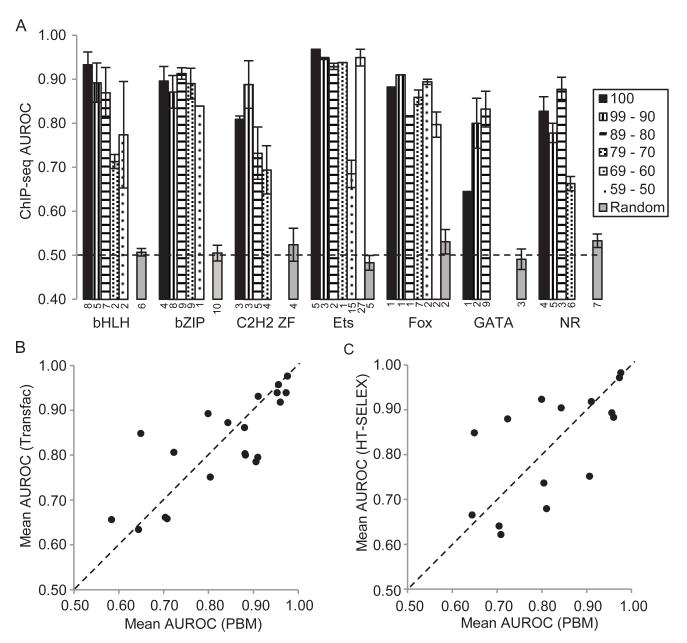
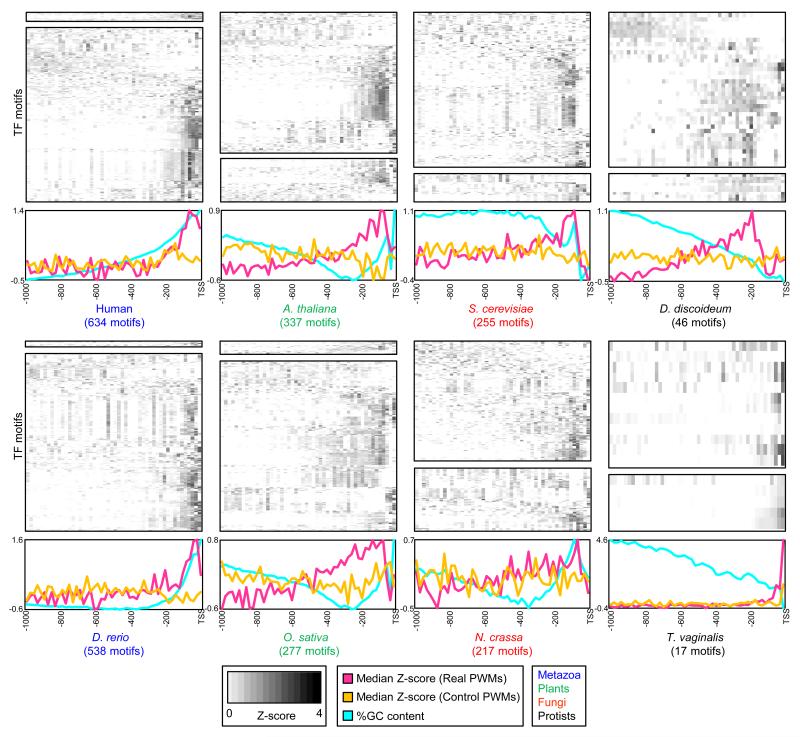
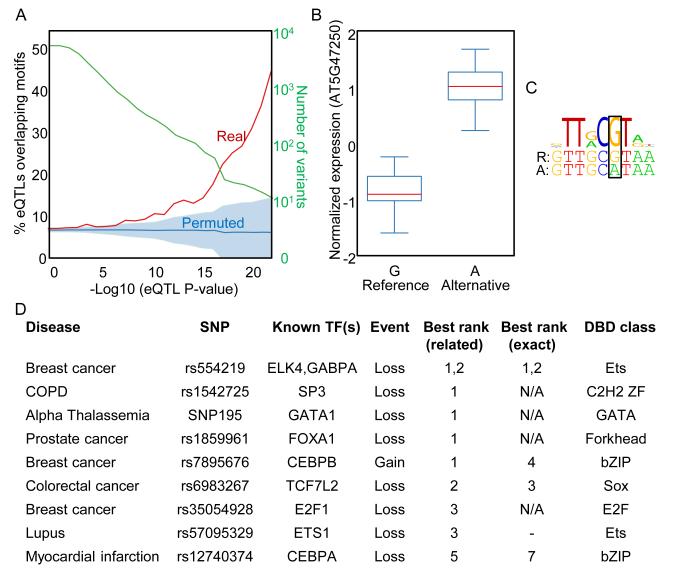
Transcription factor (TF) DNA sequence preferences direct their regulatory activity, but are currently known for only ∼1% of eukaryotic TFs. Broadly sampling DNA-binding domain (DBD) types from multiple eukaryotic clades, we determined DNA sequence preferences for >1,000 TFs encompassing 54 different DBD classes from 131 diverse eukaryotes. We find that closely related DBDs almost always have very similar DNA sequence preferences, enabling inference of motifs for ∼34% of the ∼170,000 known or predicted eukaryotic TFs. Sequences matching both measured and inferred motifs are enriched in chromatin immunoprecipitation sequencing (ChIP-seq) peaks and upstream of transcription start sites in diverse eukaryotic lineages. SNPs defining expression quantitative trait loci in Arabidopsis promoters are also enriched for predicted TF binding sites. Importantly, our motif "library" can be used to identify specific TFs whose binding may be altered by human disease risk alleles. These data present a powerful resource for mapping transcriptional networks across eukaryotes.
Copyright © 2014 Elsevier Inc. All rights reserved.
Figures
References
-
- Baldauf SL, Roger AJ, Wenk-Siefert I, Doolittle WF. A kingdom-level phylogeny of eukaryotes based on combined protein data. Science. 2000;290:972–977. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
