

A formal perturbation equation between genotype and phenotype determines the Evolutionary Action of protein-coding variations on fitness
- PMID: 25217195
- PMCID: PMC4248321
- DOI: 10.1101/gr.176214.114
A formal perturbation equation between genotype and phenotype determines the Evolutionary Action of protein-coding variations on fitness
Abstract
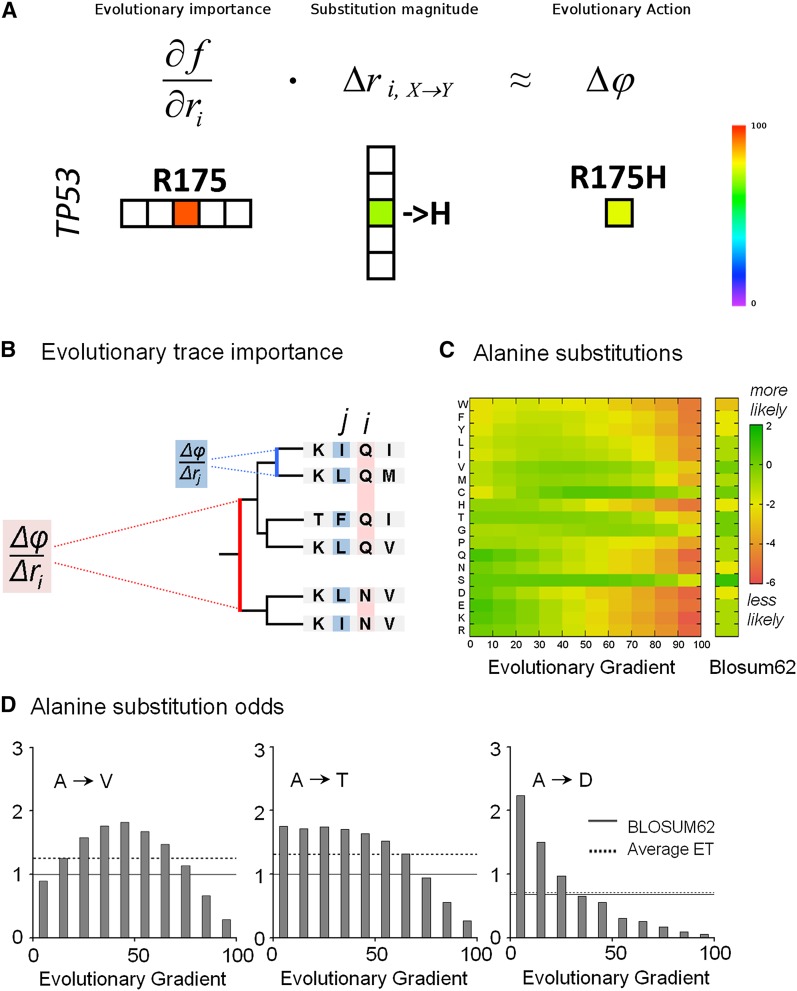
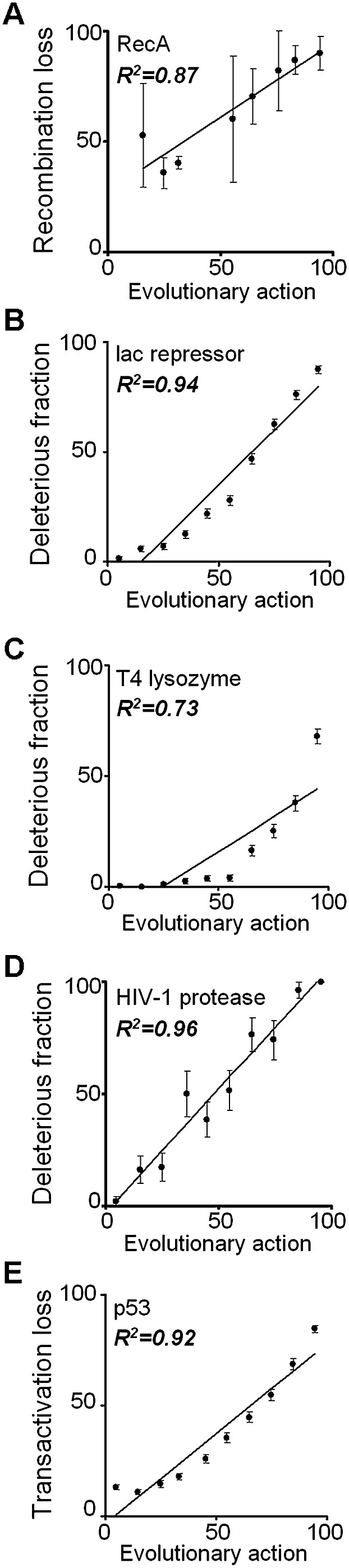
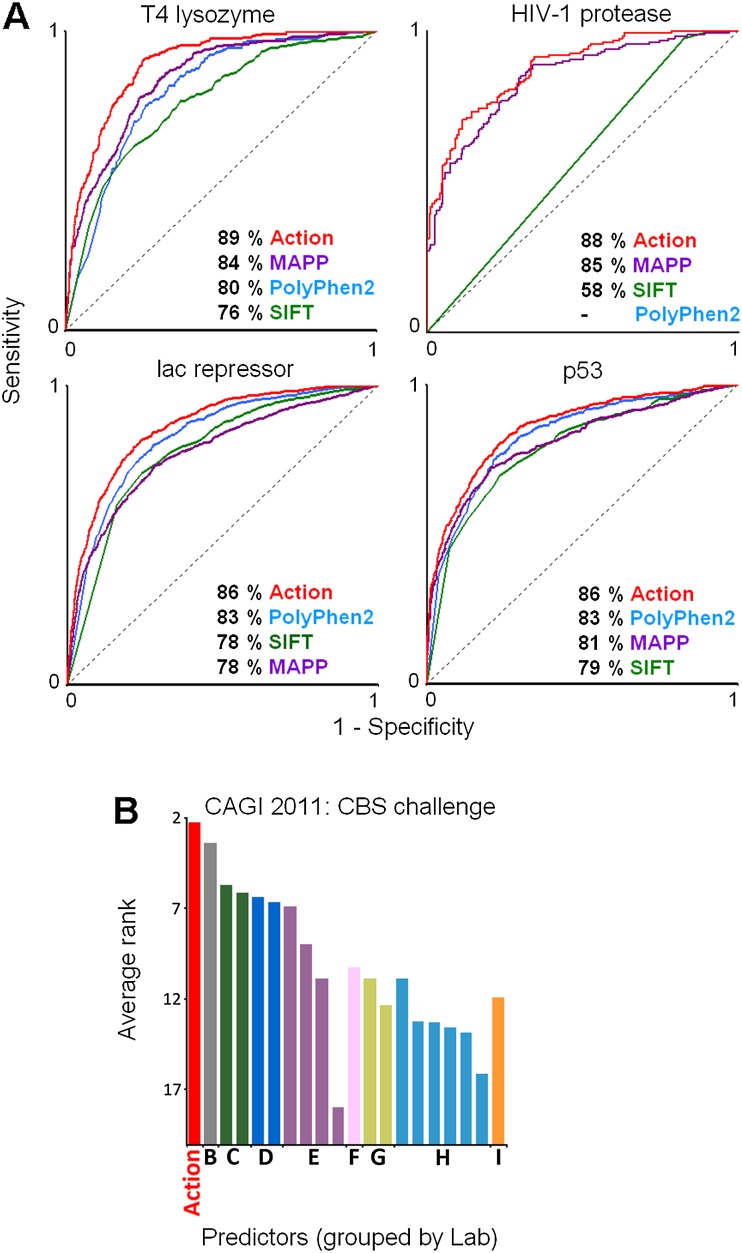
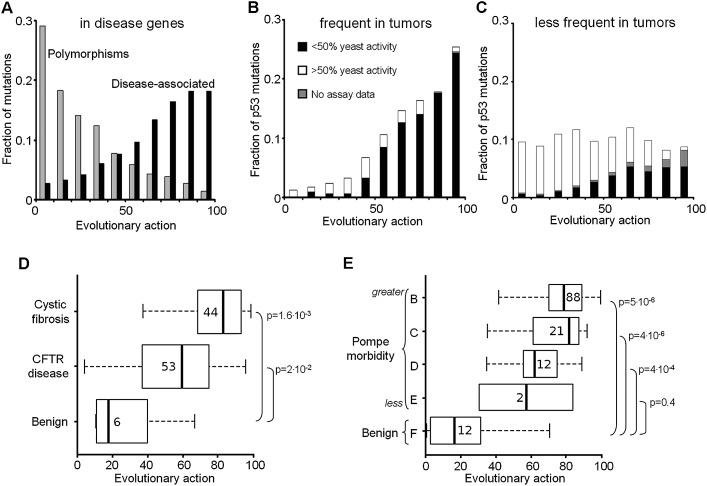
The relationship between genotype mutations and phenotype variations determines health in the short term and evolution over the long term, and it hinges on the action of mutations on fitness. A fundamental difficulty in determining this action, however, is that it depends on the unique context of each mutation, which is complex and often cryptic. As a result, the effect of most genome variations on molecular function and overall fitness remains unknown and stands apart from population genetics theories linking fitness effect to polymorphism frequency. Here, we hypothesize that evolution is a continuous and differentiable physical process coupling genotype to phenotype. This leads to a formal equation for the action of coding mutations on fitness that can be interpreted as a product of the evolutionary importance of the mutated site with the difference in amino acid similarity. Approximations for these terms are readily computable from phylogenetic sequence analysis, and we show mutational, clinical, and population genetic evidence that this action equation predicts the effect of point mutations in vivo and in vitro in diverse proteins, correlates disease-causing gene mutations with morbidity, and determines the frequency of human coding polymorphisms, respectively. Thus, elementary calculus and phylogenetics can be integrated into a perturbation analysis of the evolutionary relationship between genotype and phenotype that quantitatively links point mutations to function and fitness and that opens a new analytic framework for equations of biology. In practice, this work explicitly bridges molecular evolution with population genetics with applications from protein redesign to the clinical assessment of human genetic variations.
© 2014 Katsonis and Lichtarge; Published by Cold Spring Harbor Laboratory Press.
Figures
Similar articles
-
Objective assessment of the evolutionary action equation for the fitness effect of missense mutations across CAGI-blinded contests.Hum Mutat. 2017 Sep;38(9):1072-1084. doi: 10.1002/humu.23266. Epub 2017 Jun 21. Hum Mutat. 2017. PMID: 28544059 Free PMC article. Review.
-
CAGI5: Objective performance assessments of predictions based on the Evolutionary Action equation.Hum Mutat. 2019 Sep;40(9):1436-1454. doi: 10.1002/humu.23873. Epub 2019 Aug 7. Hum Mutat. 2019. PMID: 31317604 Free PMC article.
-
Evolution in the light of fitness landscape theory.Trends Ecol Evol. 2019 Jan;34(1):69-82. doi: 10.1016/j.tree.2018.10.009. Epub 2018 Dec 21. Trends Ecol Evol. 2019. PMID: 30583805 Review.
-
Inferring the distribution of fitness effects of spontaneous mutations in Chlamydomonas reinhardtii.PLoS Biol. 2019 Jun 26;17(6):e3000192. doi: 10.1371/journal.pbio.3000192. eCollection 2019 Jun. PLoS Biol. 2019. PMID: 31242179 Free PMC article.
-
Sampling Strategies for Experimentally Mapping Molecular Fitness Landscapes Using High-Throughput Methods.J Mol Evol. 2024 Aug;92(4):402-414. doi: 10.1007/s00239-024-10179-8. Epub 2024 Jun 17. J Mol Evol. 2024. PMID: 38886207
Cited by
-
MYC Induces Oncogenic Stress through RNA Decay and Ribonucleotide Catabolism in Breast Cancer.Cancer Discov. 2024 Sep 4;14(9):1699-1716. doi: 10.1158/2159-8290.CD-22-0649. Cancer Discov. 2024. PMID: 39193992 Free PMC article.
-
Uncovering DNA-PKcs ancient phylogeny, unique sequence motifs and insights for human disease.Prog Biophys Mol Biol. 2021 Aug;163:87-108. doi: 10.1016/j.pbiomolbio.2020.09.010. Epub 2020 Oct 6. Prog Biophys Mol Biol. 2021. PMID: 33035590 Free PMC article.
-
Predicting pathogenicity of missense variants with weakly supervised regression.Hum Mutat. 2019 Sep;40(9):1579-1592. doi: 10.1002/humu.23826. Epub 2019 Aug 7. Hum Mutat. 2019. PMID: 31144781 Free PMC article.
-
Deleterious Effect of RAS and Evolutionary High-risk TP53 Double Mutation in Colorectal Liver Metastases.Ann Surg. 2019 May;269(5):917-923. doi: 10.1097/SLA.0000000000002450. Ann Surg. 2019. PMID: 28767562 Free PMC article.
-
DISCOVERY OF FUNCTIONAL AND DISEASE PATHWAYS BY COMMUNITY DETECTION IN PROTEIN-PROTEIN INTERACTION NETWORKS.Pac Symp Biocomput. 2017;22:336-347. doi: 10.1142/9789813207813_0032. Pac Symp Biocomput. 2017. PMID: 27896987 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials