

Prioritizing genes for X-linked diseases using population exome data
- PMID: 25217573
- PMCID: PMC4291241
- DOI: 10.1093/hmg/ddu473
Prioritizing genes for X-linked diseases using population exome data
Abstract
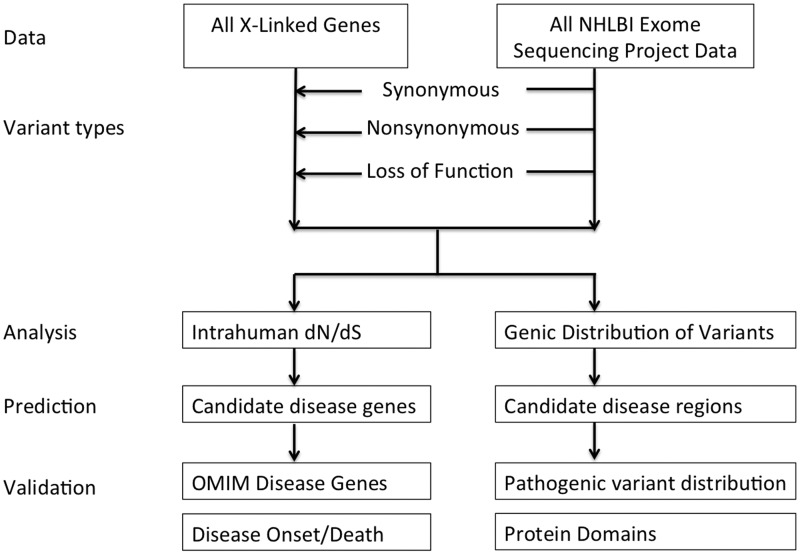
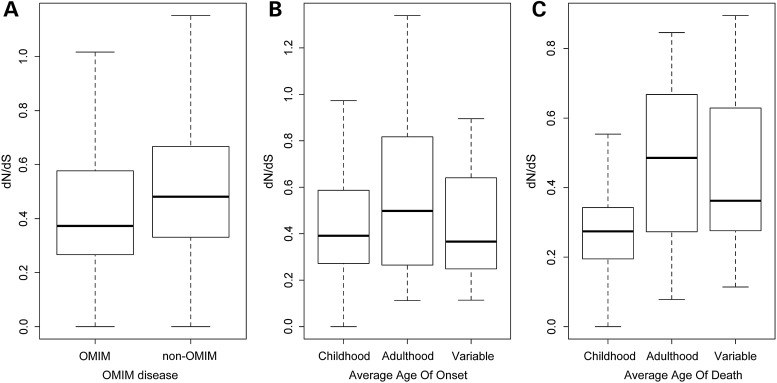
Many new disease genes can be identified through high-throughput sequencing. Yet, variant interpretation for the large amounts of genomic data remains a challenge given variation of uncertain significance and genes that lack disease annotation. As clinically significant disease genes may be subject to negative selection, we developed a prediction method that measures paucity of non-synonymous variation in the human population to infer gene-based pathogenicity. Integrating human exome data of over 6000 individuals from the NHLBI Exome Sequencing Project, we tested the utility of the prediction method based on the ratio of non-synonymous to synonymous substitution rates (dN/dS) on X-chromosome genes. A low dN/dS ratio characterized genes associated with childhood disease and outcome. Furthermore, we identify new candidates for diseases with early mortality and demonstrate intragenic localized patterns of variants that suggest pathogenic hotspots. Our results suggest that intrahuman substitution analysis is a valuable tool to help prioritize novel disease genes in sequence interpretation.
© The Author 2014. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures


References
-
- Xue Y., Chen Y., Ayub Q., Huang N., Ball E.V., Mort M., Phillips A.D., Shaw K., Stenson P.D., Cooper D.N., et al. Deleterious- and disease-allele prevalence in healthy individuals: insights from current predictions, mutation databases, and population-scale resequencing. Am. J. Hum. Genet. 2012;91:1022–1032. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
