

CNS, lung, and lymph node involvement in Gaucher disease type 3 after 11 years of therapy: clinical, histopathologic, and biochemical findings
- PMID: 25219293
- PMCID: PMC4312736
- DOI: 10.1016/j.ymgme.2014.08.011
CNS, lung, and lymph node involvement in Gaucher disease type 3 after 11 years of therapy: clinical, histopathologic, and biochemical findings
Abstract
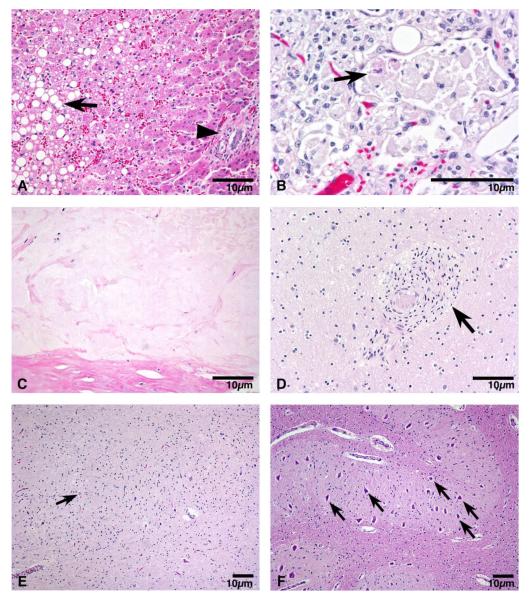
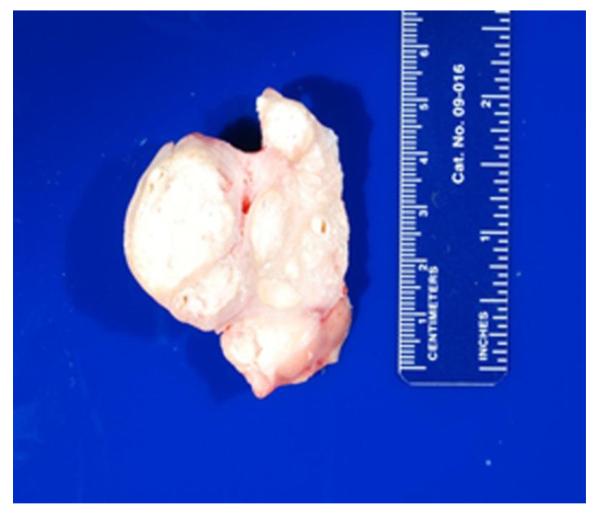
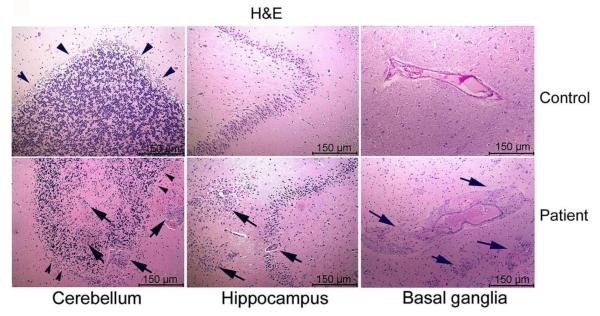
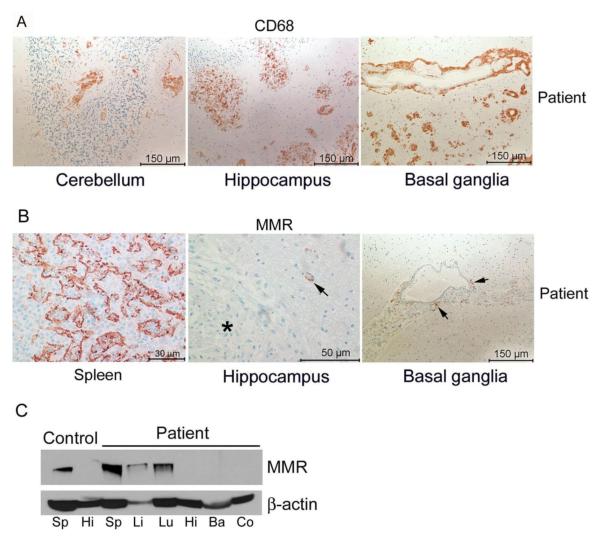
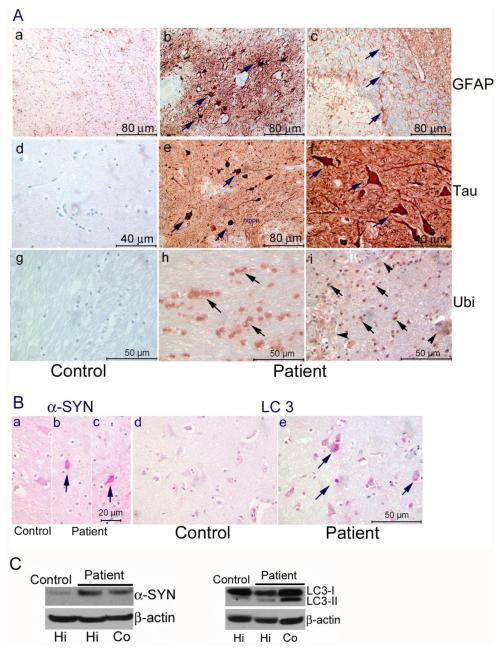
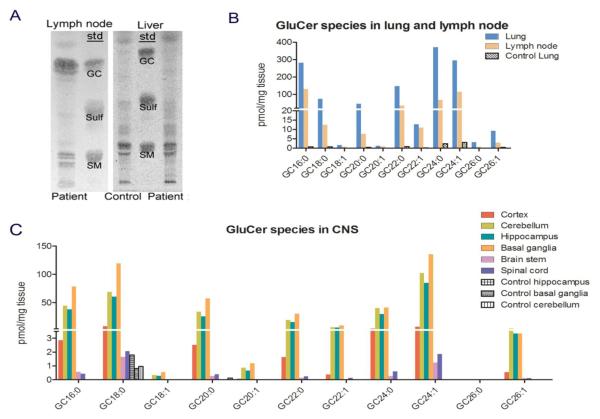
A Caucasian male with Gaucher disease type 3, treated with continuous enzyme therapy (ET) for 11 years, experienced progressive mesenteric and retroperitoneal lymphadenopathy, lung disease, and neurological involvement leading to death at an age of 12.5 years. Autopsy showed significant pathology of the brain, lymph nodes, and lungs. Liver and spleen glucosylceramide (GluCer) and glucosylsphingosine (GluS) levels were nearly normal and storage cells were cleared. Clusters of macrophages and very elevated GluCer and GluS levels were in the lungs, and brain parenchymal and perivascular regions. Compared to normal brain GluCer (GC 18:0), GluCer species with long fatty acid acyl chains were increased in the patient's brain. This profile was similar to that in the patient's lungs, suggesting that these lipids were present in brain perivascular macrophages. In the patient's brain, generalized astrogliosis, and enhanced LC3, ubiquitin, and Tau signals were identified in the regions surrounding macrophage clusters, indicating proinflammation, altered autophagy, and neurodegeneration. These findings highlight the altered phenotypes resulting from increased longevity due to ET, as well as those in poorly accessible compartments of brain and lung, which manifested progressive disease involvement despite ET.
Keywords: Enzyme replacement therapy; Gaucher disease; Lymphadenopathy; Lysosomal storage disease; Pathology.
Copyright © 2014 Elsevier Inc. All rights reserved.
Figures
References
-
- Grabowski G, Petsko G, Kolodny E. Gaucher Disease. In: Valle D, Beaudet A, Vogelstein B, Kinzler K, Antonarakis S, Ballabio A, Gibson K, Mitchell G, editors. The Online Metabolic and Molecular Bases of Inherited Disease. The McGraw-Hill Companies, Inc.; New York, NY: [Accessed on; 06/23/14]. 2010. Chapt 146.
-
- Altarescu G, Hill S, Wiggs E, Jeffries N, Kreps C, Parker CC, Brady RO, Barton NW, Schiffmann R. The efficacy of enzyme replacement therapy in patients with chronic neuronopathic Gaucher’s disease. J Pediatr. 2001;138:539–547. - PubMed
-
- Burrow TA, Cohen MB, Bokulic R, Deutsch G, Choudhary A, Falcone RA, Jr., Grabowski GA. Gaucher disease: progressive mesenteric and mediastinal lymphadenopathy despite enzyme therapy. J Pediatr. 2007;150:202–206. - PubMed
-
- Grabowski GA, Leslie N, Wenstrup R. Enzyme therapy for Gaucher disease: the first 5 years. Blood Rev. 1998;12:115–133. - PubMed
-
- Zhao H, Bailey LA, Elsas LJ, 2nd, Grinzaid KA, Grabowski GA. Gaucher disease: in vivo evidence for allele dose leading to neuronopathic and nonneuronopathic phenotypes. Am J Med Genet A. 2003;116A:52–56. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous