

The genetic signatures of pediatric high-grade glioma: no longer a one-act play
- PMID: 25219808
- PMCID: PMC4170681
- DOI: 10.1016/j.semradonc.2014.06.003
The genetic signatures of pediatric high-grade glioma: no longer a one-act play
Abstract
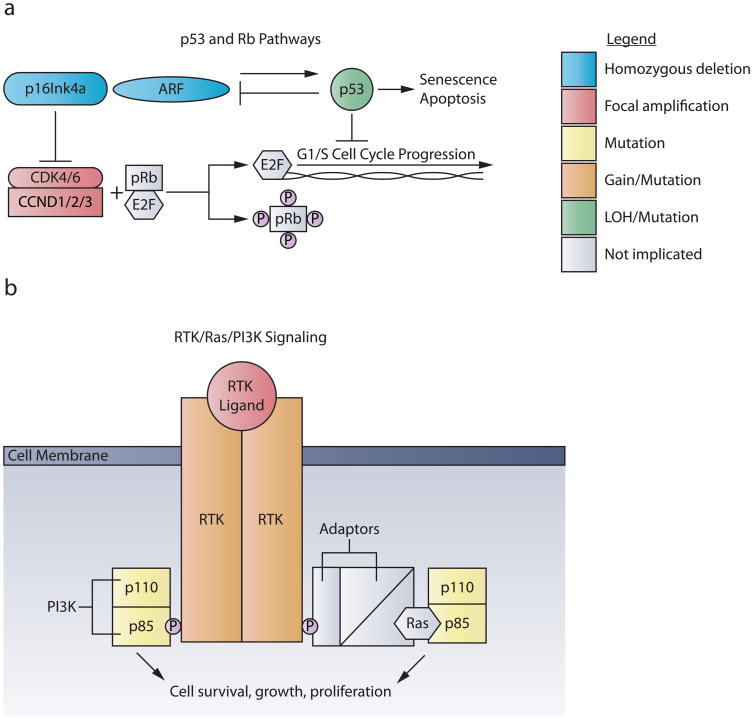
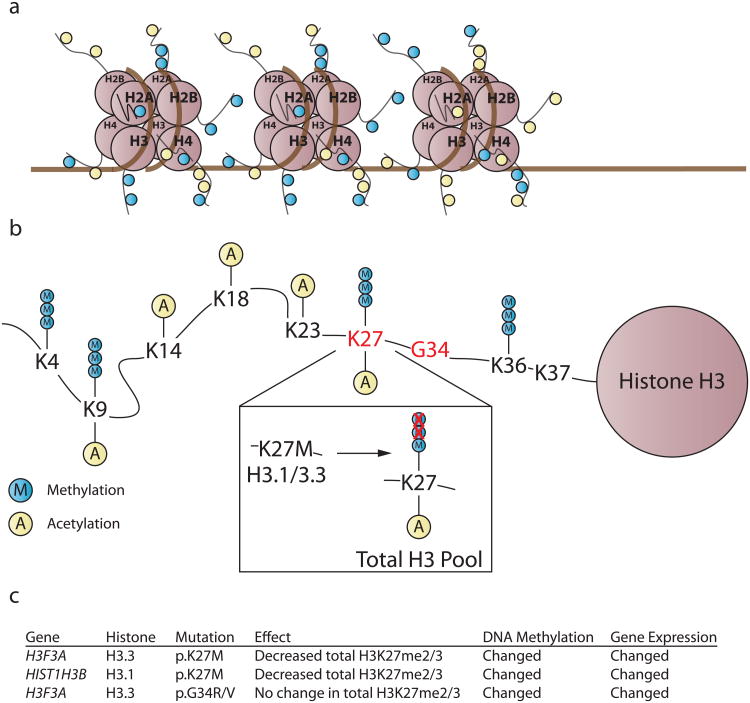
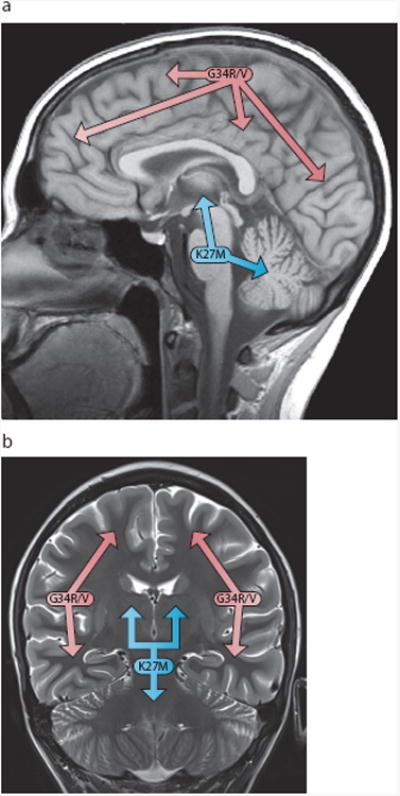
Advances in understanding pediatric high-grade glioma (pHGG) genetics have revealed key differences between pHGG and adult HGG and have uncovered unique molecular drivers among subgroups within pHGG. The 3 core adult HGG pathways, the receptor tyrosine kinase-Ras-phosphatidylinositide 3-kinase, p53, and retinoblastoma networks, are also disrupted in pHGG, but they exhibit a different spectrum of effectors targeted by mutation. There are also similarities and differences in the genomic landscape of diffuse intrinsic pontine glioma (DIPG) and pediatric nonbrainstem (pNBS)-HGG. In 2012, histone H3 mutations were identified in nearly 80% of DIPGs and ~35% of pNBS-HGG. These were the first reports of histone mutations in human cancer, implicating novel biology in pediatric gliomagenesis. Additionally, DIPG and midline pNBS-HGG vary in the frequency and specific histone H3 amino acid substitution compared with pNBS-HGGs arising in the cerebral hemispheres, demonstrating a molecular difference among pHGG subgroups. The gene expression signatures as well as DNA methylation signatures of these tumors are also distinctive, reflecting a combination of the driving mutations and the developmental context from which they arise. These data collectively highlight unique selective pressures within the developing brainstem and solidify DIPG as a specific molecular and biological entity among pHGGs. Emerging studies continue to identify novel mutations that distinguish subgroups of pHGG. The molecular heterogeneity among pHGGs will undoubtedly have clinical implications moving forward. The discovery of unique oncogenic drivers is a critical first step in providing patients with appropriate, targeted therapies. Despite these insights, our vantage point has been largely limited to an in-depth analysis of protein coding sequences. Given the clear importance of histone mutations in pHGG, it will be interesting to see how aberrant epigenetic regulation contributes to tumorigenesis in the pediatric context. New mechanistic insights may allow for the identification of distinct vulnerabilities in this devastating spectrum of childhood tumors.
Copyright © 2014 Elsevier Inc. All rights reserved.
Conflict of interest statement
No potential conflicts of interest were disclosed.
Figures
Similar articles
-
Oncohistones and disrupted development in pediatric-type diffuse high-grade glioma.Cancer Metastasis Rev. 2023 Jun;42(2):367-388. doi: 10.1007/s10555-023-10105-2. Epub 2023 Apr 29. Cancer Metastasis Rev. 2023. PMID: 37119408 Free PMC article. Review.
-
Hemispherical Pediatric High-Grade Glioma: Molecular Basis and Therapeutic Opportunities.Int J Mol Sci. 2020 Dec 17;21(24):9654. doi: 10.3390/ijms21249654. Int J Mol Sci. 2020. PMID: 33348922 Free PMC article. Review.
-
Novel oncogenic PDGFRA mutations in pediatric high-grade gliomas.Cancer Res. 2013 Oct 15;73(20):6219-29. doi: 10.1158/0008-5472.CAN-13-1491. Epub 2013 Aug 22. Cancer Res. 2013. PMID: 23970477 Free PMC article.
-
Unique genetic and epigenetic mechanisms driving paediatric diffuse high-grade glioma.Nat Rev Cancer. 2014 Oct;14(10):10.1038/nrc3811. doi: 10.1038/nrc3811. Nat Rev Cancer. 2014. PMID: 25230881 Free PMC article. Review.
-
Characterizing temporal genomic heterogeneity in pediatric high-grade gliomas.Acta Neuropathol Commun. 2017 Oct 30;5(1):78. doi: 10.1186/s40478-017-0479-8. Acta Neuropathol Commun. 2017. PMID: 29084603 Free PMC article.
Cited by
-
An overview of neuro-oncology research and practice in Iran, three years with the NOSC initiative.Int J Clin Exp Med. 2015 Mar 15;8(3):3946-55. eCollection 2015. Int J Clin Exp Med. 2015. PMID: 26064296 Free PMC article.
-
WHO grade has no prognostic value in the pediatric high-grade glioma included in the HERBY trial.Neuro Oncol. 2020 Jan 11;22(1):116-127. doi: 10.1093/neuonc/noz142. Neuro Oncol. 2020. PMID: 31419298 Free PMC article.
-
NKG2D ligand expression in pediatric brain tumors.Cancer Biol Ther. 2016 Dec;17(12):1253-1265. doi: 10.1080/15384047.2016.1250047. Epub 2016 Nov 11. Cancer Biol Ther. 2016. PMID: 27834580 Free PMC article.
-
Modulating autophagy as a therapeutic strategy for the treatment of paediatric high-grade glioma.Brain Pathol. 2019 Nov;29(6):707-725. doi: 10.1111/bpa.12729. Epub 2019 May 13. Brain Pathol. 2019. PMID: 31012506 Free PMC article. Review.
-
Brain Tumor Biobank Development for Precision Medicine: Role of the Neurosurgeon.Front Oncol. 2021 Apr 26;11:662260. doi: 10.3389/fonc.2021.662260. eCollection 2021. Front Oncol. 2021. PMID: 33981610 Free PMC article.
References
-
- Broniscer A, Gajjar A. Supratentorial high-grade astrocytoma and diffuse brainstem glioma: two challenges for the pediatric oncologist. Oncologist. 2004;9:197–206. - PubMed
-
- Chassot A, et al. Radiotherapy with concurrent and adjuvant temozolomide in children with newly diagnosed diffuse intrinsic pontine glioma. J Neurooncol. 2012;106:399–407. - PubMed
-
- Stupp R, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–96. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
