

Characterizing the pocketome of Mycobacterium tuberculosis and application in rationalizing polypharmacological target selection
- PMID: 25220818
- PMCID: PMC5376175
- DOI: 10.1038/srep06356
Characterizing the pocketome of Mycobacterium tuberculosis and application in rationalizing polypharmacological target selection
Abstract
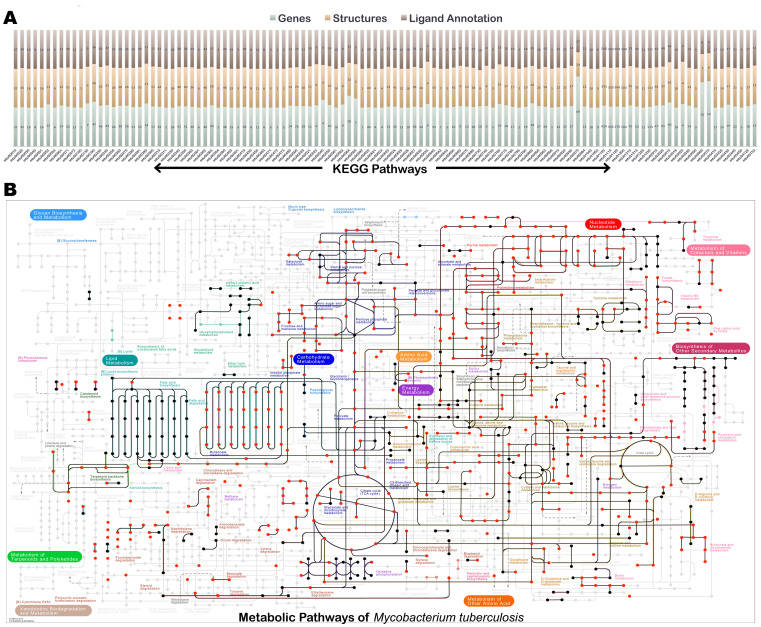
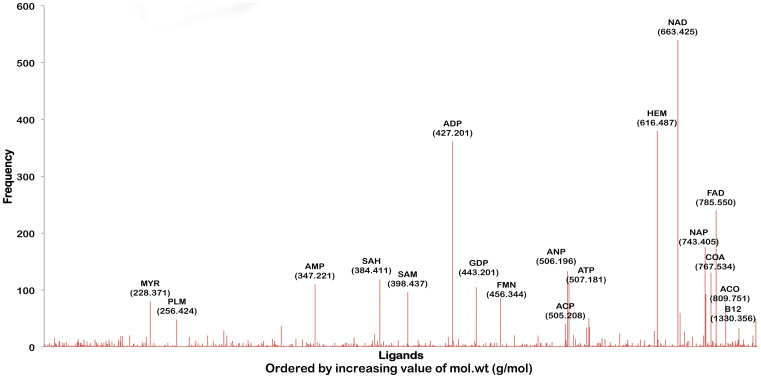
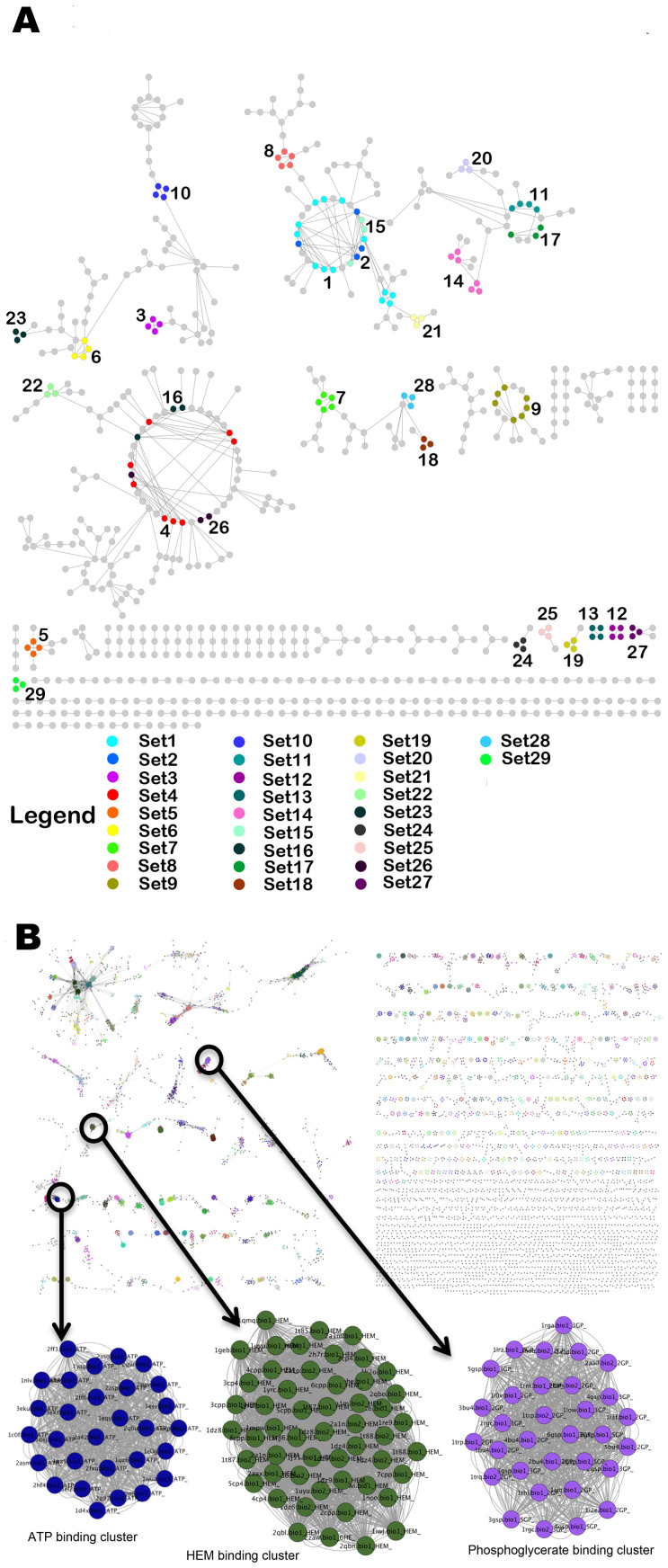
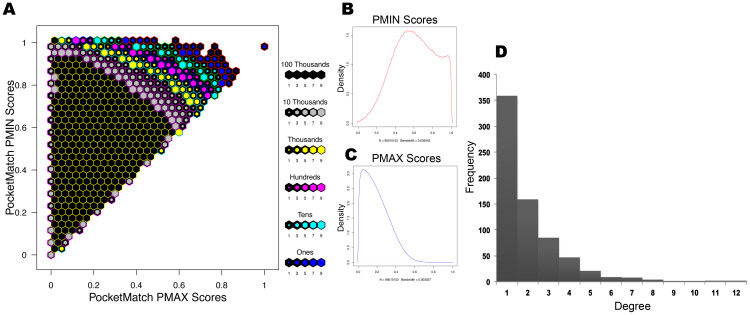
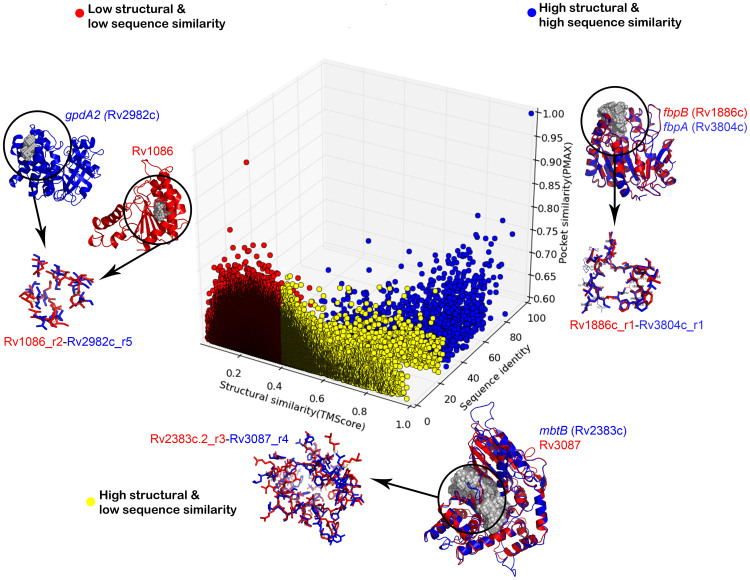
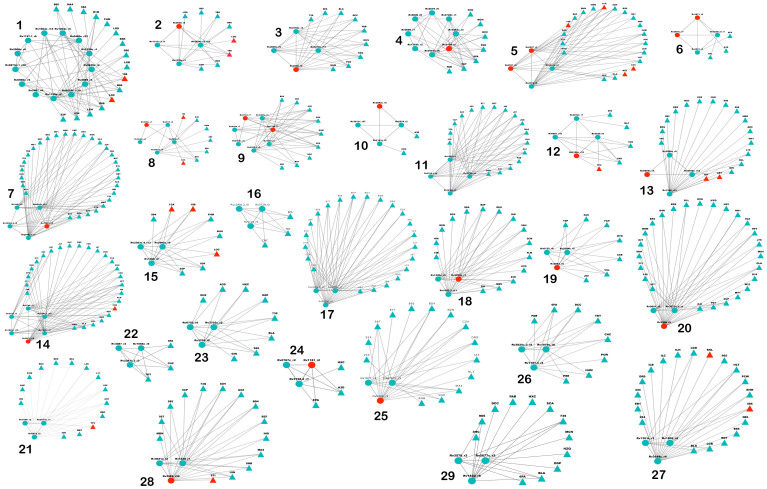
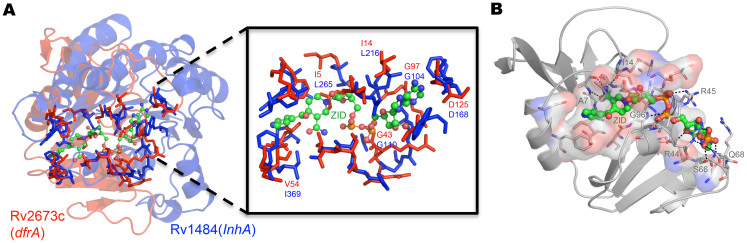
Polypharmacology is beginning to emerge as an important concept in the field of drug discovery. However, there are no established approaches to either select appropriate target sets or design polypharmacological drugs. Here, we propose a structural-proteomics approach that utilizes the structural information of the binding sites at a genome-scale obtained through in-house algorithms to characterize the pocketome, yielding a list of ligands that can participate in various biochemical events in the mycobacterial cell. The pocket-type space is seen to be much larger than the sequence or fold-space, suggesting that variations at the site-level contribute significantly to functional repertoire of the organism. All-pair comparisons of binding sites within Mycobacterium tuberculosis (Mtb), pocket-similarity network construction and clustering result in identification of binding-site sets, each containing a group of similar binding sites, theoretically having a potential to interact with a common set of compounds. A polypharmacology index is formulated to rank targets by incorporating a measure of druggability and similarity to other pockets within the proteome. This study presents a rational approach to identify targets with polypharmacological potential along with possible drugs for repurposing, while simultaneously, obtaining clues on lead compounds for use in new drug-discovery pipelines.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
-
- Konopa K. & Jassem J. The role of pemetrexed combined with targeted agents for non-small cell lung cancer. Curr Drug Targets 11, 2–11 (2010). - PubMed
-
- Winum J. Y., Maresca A., Carta F., Scozzafava A. & Supuran C. T. Polypharmacology of sulfonamides: pazopanib, a multitargeted receptor tyrosine kinase inhibitor in clinical use, potently inhibits several mammalian carbonic anhydrases. Chem Commun (Camb) 48, 8177–9 (2012). - PubMed
-
- Hopkins A. L. Network pharmacology: the next paradigm in drug discovery. Nat Chem Biol 4, 682–90 (2008). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
