

Cholesterol in brain disease: sometimes determinant and frequently implicated
- PMID: 25223281
- PMCID: PMC4253844
- DOI: 10.15252/embr.201439225
Cholesterol in brain disease: sometimes determinant and frequently implicated
Abstract
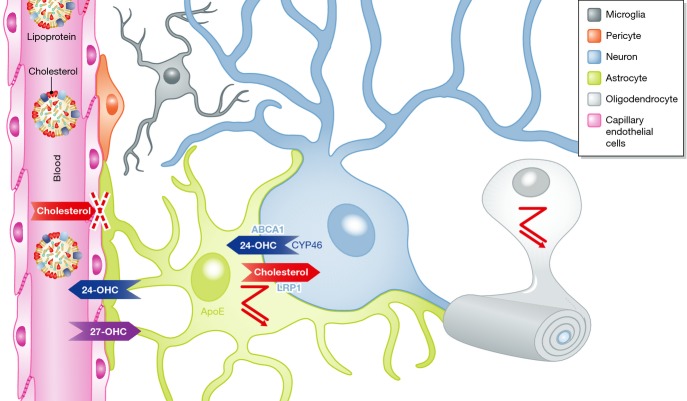
Cholesterol is essential for neuronal physiology, both during development and in the adult life: as a major component of cell membranes and precursor of steroid hormones, it contributes to the regulation of ion permeability, cell shape, cell-cell interaction, and transmembrane signaling. Consistently, hereditary diseases with mutations in cholesterol-related genes result in impaired brain function during early life. In addition, defects in brain cholesterol metabolism may contribute to neurological syndromes, such as Alzheimer's disease (AD), Huntington's disease (HD), and Parkinson's disease (PD), and even to the cognitive deficits typical of the old age. In these cases, brain cholesterol defects may be secondary to disease-causing elements and contribute to the functional deficits by altering synaptic functions. In the first part of this review, we will describe hereditary and non-hereditary causes of cholesterol dyshomeostasis and the relationship to brain diseases. In the second part, we will focus on the mechanisms by which perturbation of cholesterol metabolism can affect synaptic function.
Keywords: brain disease; cholesterol metabolism; cognition.
© 2014 The Authors.
Figures



References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
