

Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations
- PMID: 25223734
- PMCID: PMC4264969
- DOI: 10.1158/2159-8290.CD-14-0622
Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations
Abstract
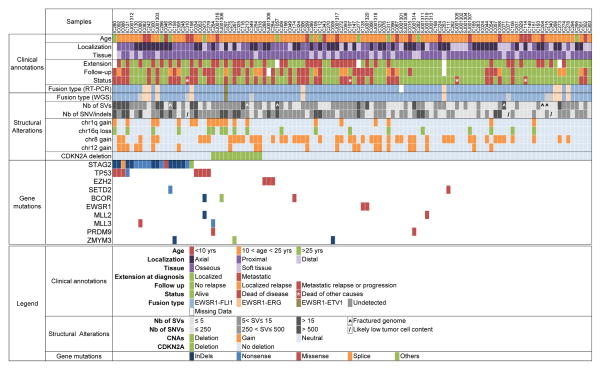
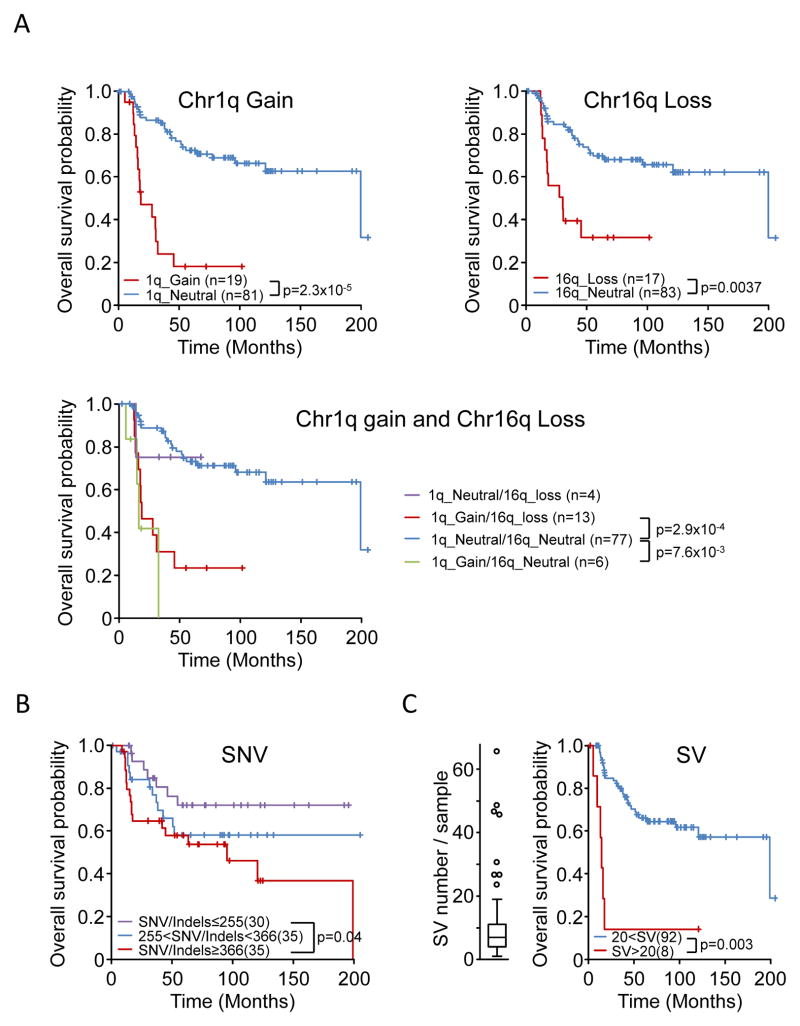
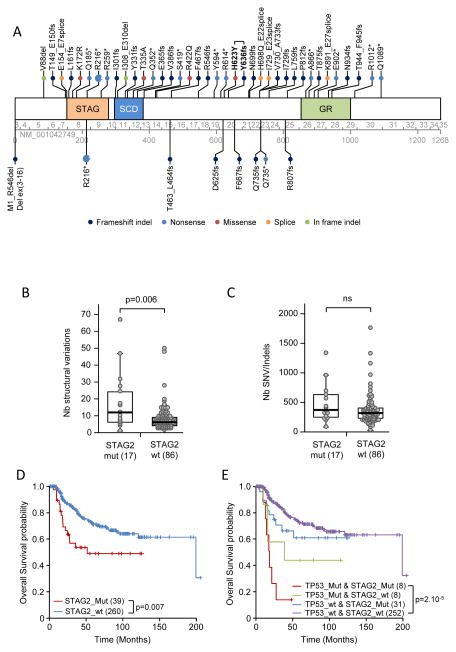
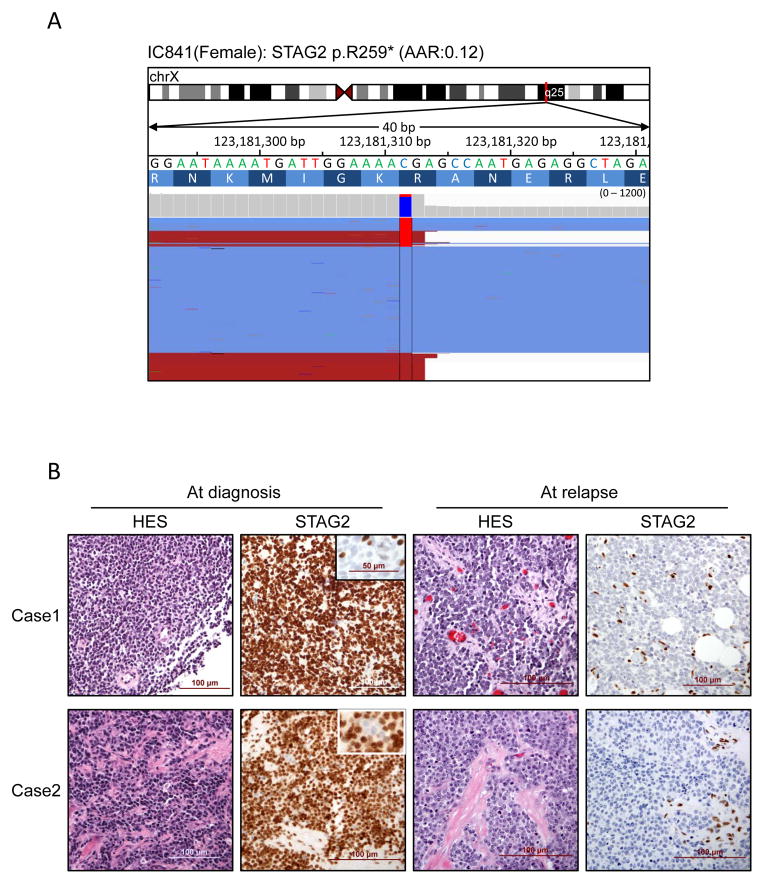
Ewing sarcoma is a primary bone tumor initiated by EWSR1-ETS gene fusions. To identify secondary genetic lesions that contribute to tumor progression, we performed whole-genome sequencing of 112 Ewing sarcoma samples and matched germline DNA. Overall, Ewing sarcoma tumors had relatively few single-nucleotide variants, indels, structural variants, and copy-number alterations. Apart from whole chromosome arm copy-number changes, the most common somatic mutations were detected in STAG2 (17%), CDKN2A (12%), TP53 (7%), EZH2, BCOR, and ZMYM3 (2.7% each). Strikingly, STAG2 mutations and CDKN2A deletions were mutually exclusive, as confirmed in Ewing sarcoma cell lines. In an expanded cohort of 299 patients with clinical data, we discovered that STAG2 and TP53 mutations are often concurrent and are associated with poor outcome. Finally, we detected subclonal STAG2 mutations in diagnostic tumors and expansion of STAG2-immunonegative cells in relapsed tumors as compared with matched diagnostic samples.
Significance: Whole-genome sequencing reveals that the somatic mutation rate in Ewing sarcoma is low. Tumors that harbor STAG2 and TP53 mutations have a particularly dismal prognosis with current treatments and require alternative therapies. Novel drugs that target epigenetic regulators may constitute viable therapeutic strategies in a subset of patients with mutations in chromatin modifiers.
©2014 American Association for Cancer Research.
Conflict of interest statement
Figures
References
-
- Delattre O, Zucman J, Plougastel B, Desmaze C, Melot T, Peter M, et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature. 1992;359:162–5. - PubMed
-
- Brisset S, Schleiermacher G, Peter M, Mairal A, Oberlin O, Delattre O, et al. CGH analysis of secondary genetic changes in Ewing tumors: correlation with metastatic disease in a series of 43 cases. Cancer Genet Cytogenet. 2001;130:57–61. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
