

Population and single-cell genomics reveal the Aire dependency, relief from Polycomb silencing, and distribution of self-antigen expression in thymic epithelia
- PMID: 25224068
- PMCID: PMC4248310
- DOI: 10.1101/gr.171645.113
Population and single-cell genomics reveal the Aire dependency, relief from Polycomb silencing, and distribution of self-antigen expression in thymic epithelia
Abstract
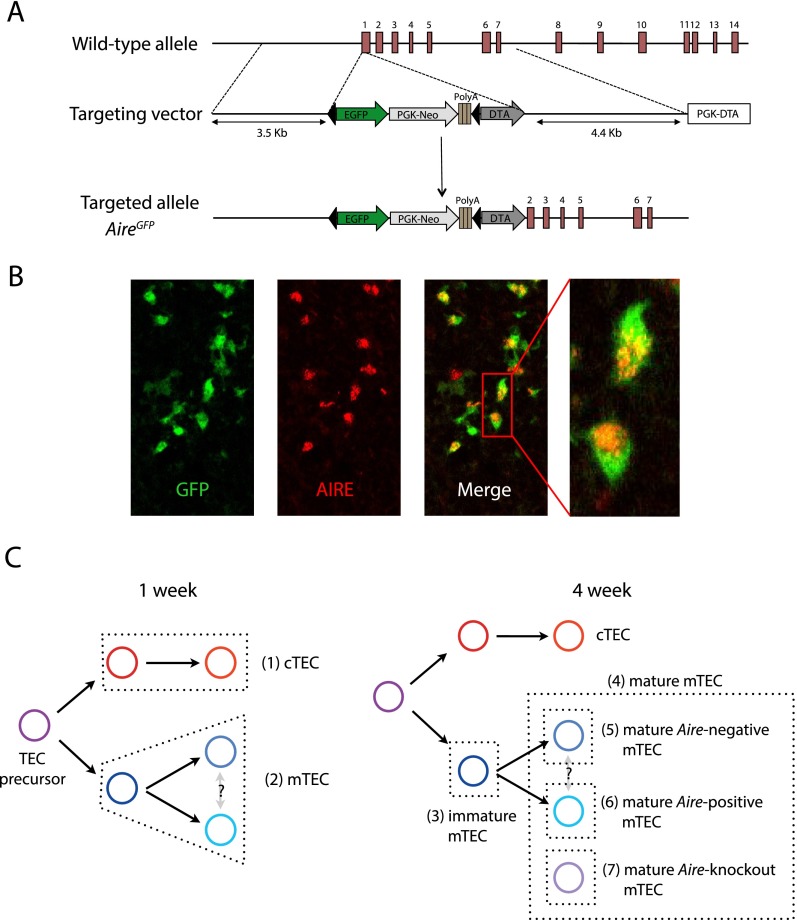
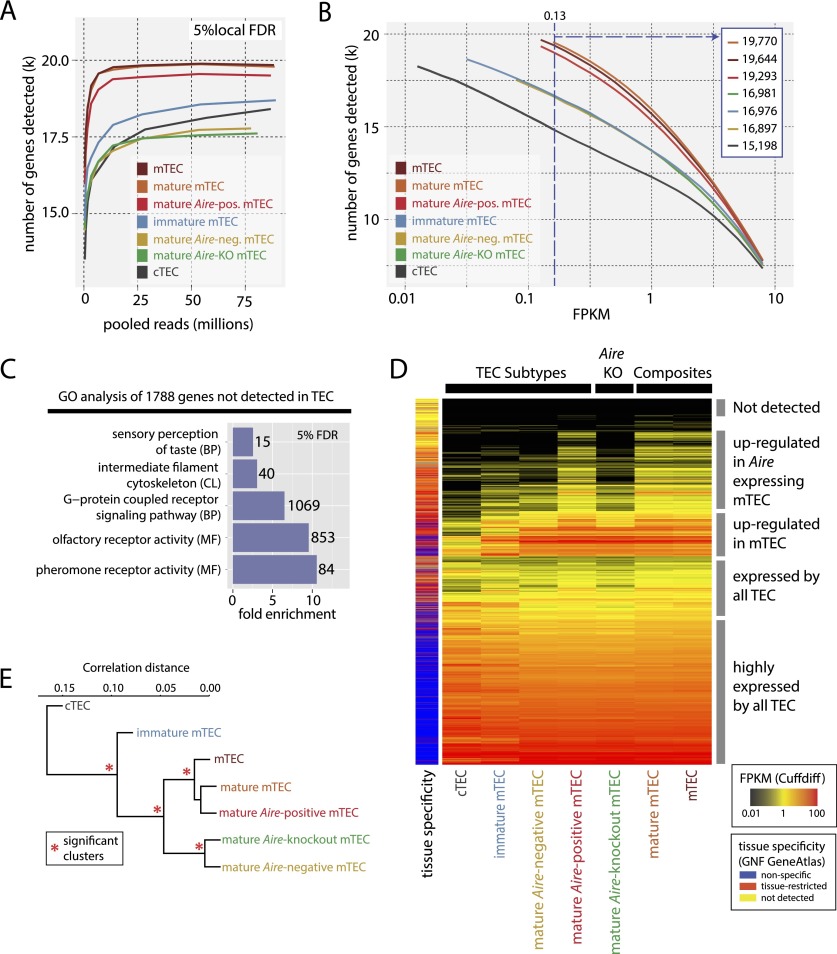
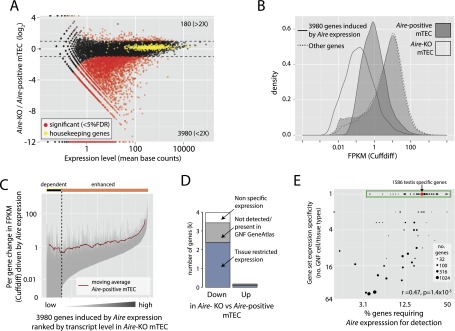
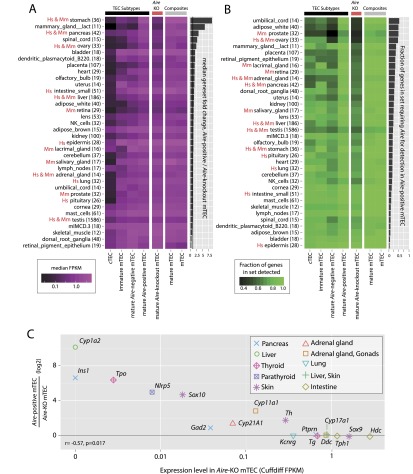
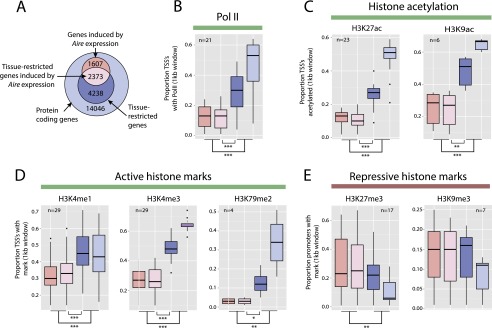
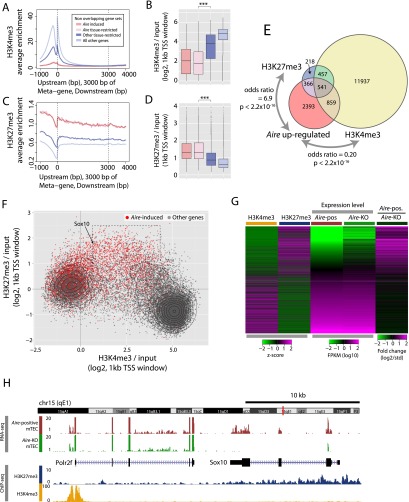
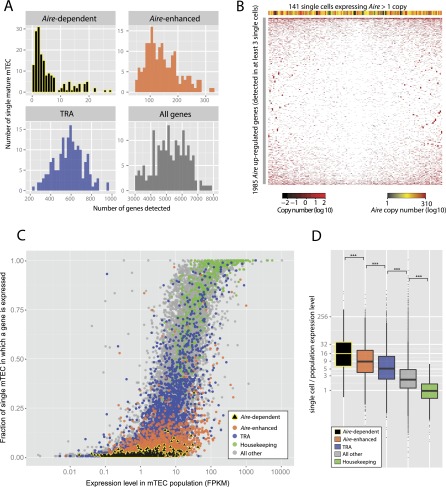
Promiscuous gene expression (PGE) by thymic epithelial cells (TEC) is essential for generating a diverse T cell antigen receptor repertoire tolerant to self-antigens, and thus for avoiding autoimmunity. Nevertheless, the extent and nature of this unusual expression program within TEC populations and single cells are unknown. Using deep transcriptome sequencing of carefully identified mouse TEC subpopulations, we discovered a program of PGE that is common between medullary (m) and cortical TEC, further elaborated in mTEC, and completed in mature mTEC expressing the autoimmune regulator gene (Aire). TEC populations are capable of expressing up to 19,293 protein-coding genes, the highest number of genes known to be expressed in any cell type. Remarkably, in mouse mTEC, Aire expression alone positively regulates 3980 tissue-restricted genes. Notably, the tissue specificities of these genes include known targets of autoimmunity in human AIRE deficiency. Led by the observation that genes induced by Aire expression are generally characterized by a repressive chromatin state in somatic tissues, we found these genes to be strongly associated with H3K27me3 marks in mTEC. Our findings are consistent with AIRE targeting and inducing the promiscuous expression of genes previously epigenetically silenced by Polycomb group proteins. Comparison of the transcriptomes of 174 single mTEC indicates that genes induced by Aire expression are transcribed stochastically at low cell frequency. Furthermore, when present, Aire expression-dependent transcript levels were 16-fold higher, on average, in individual TEC than in the mTEC population.
© 2014 Sansom et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Abramson J, Giraud M, Benoist C, Mathis D. 2010. Aire’s partners in the molecular control of immunological tolerance. Cell 140: 123–135. - PubMed
-
- Alfonso R, Lutz T, Rodriguez A, Chavez JP, Rodriguez P, Gutierrez S, Nieto A. 2011. CHD6 chromatin remodeler is a negative modulator of influenza virus replication that relocates to inactive chromatin upon infection. Cell Microbiol 13: 1894–1906. - PubMed
-
- Aloia L, Di Stefano B, Di Croce L. 2013. Polycomb complexes in stem cells and embryonic development. Development 140: 2525–2534. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases