

Unique genomic arrangements in an invasive serotype M23 strain of Streptococcus pyogenes identify genes that induce hypervirulence
- PMID: 25225265
- PMCID: PMC4248872
- DOI: 10.1128/JB.02131-14
Unique genomic arrangements in an invasive serotype M23 strain of Streptococcus pyogenes identify genes that induce hypervirulence
Abstract
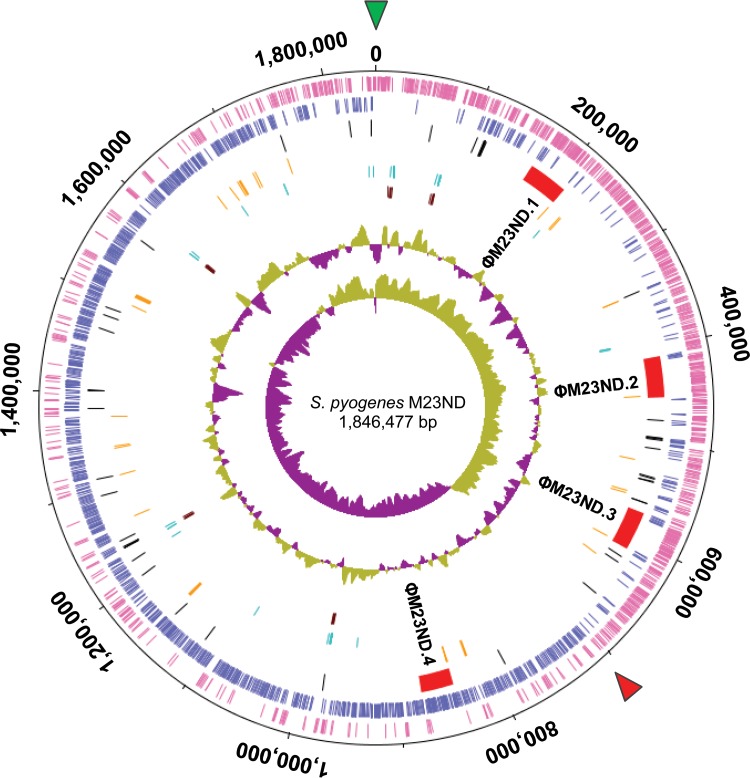
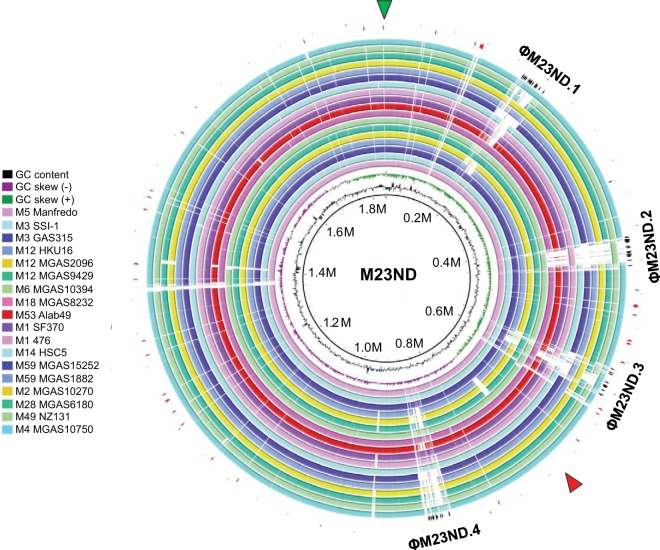
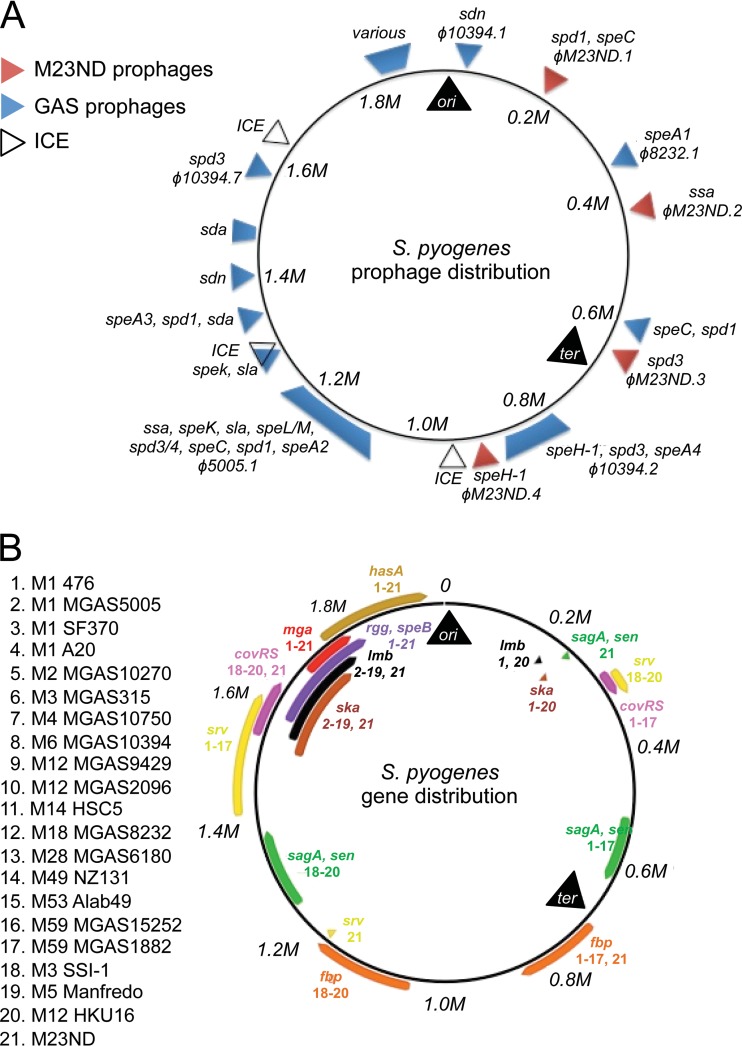
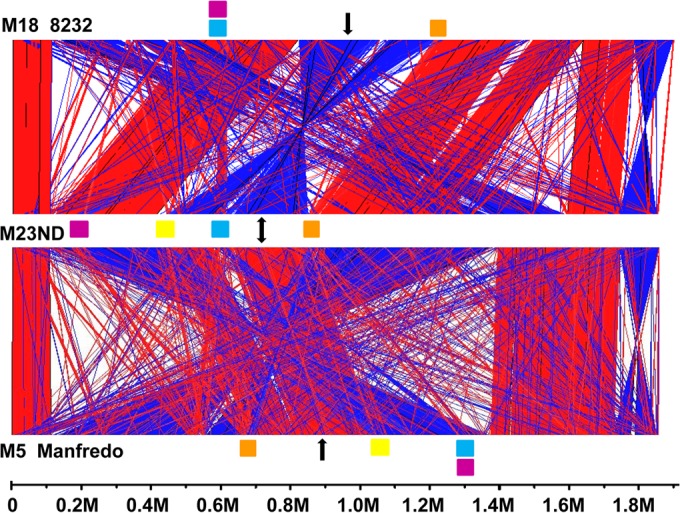
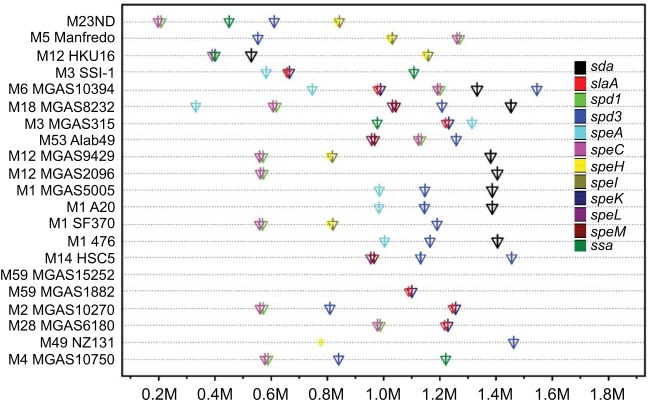
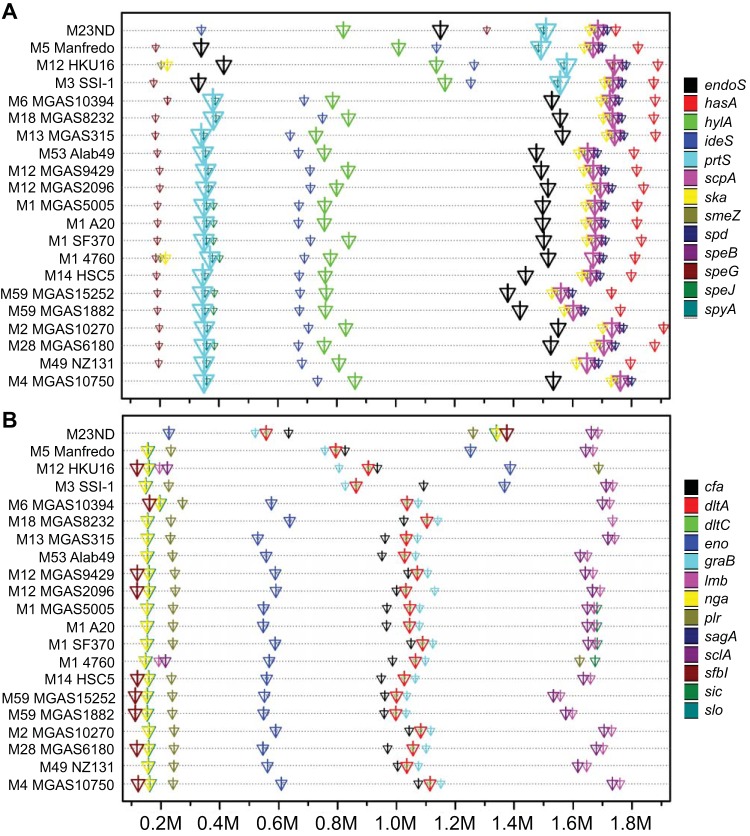
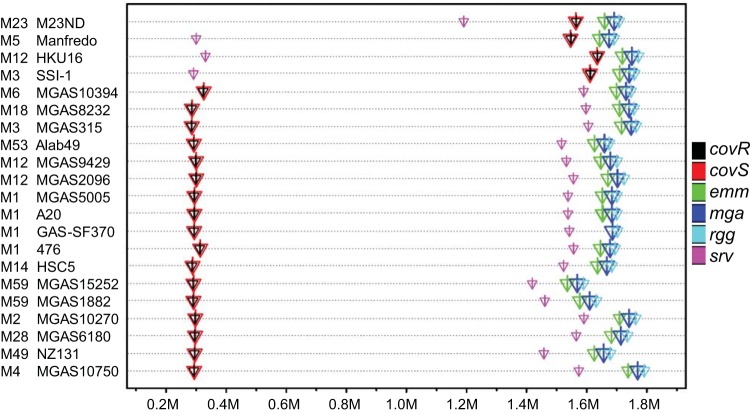
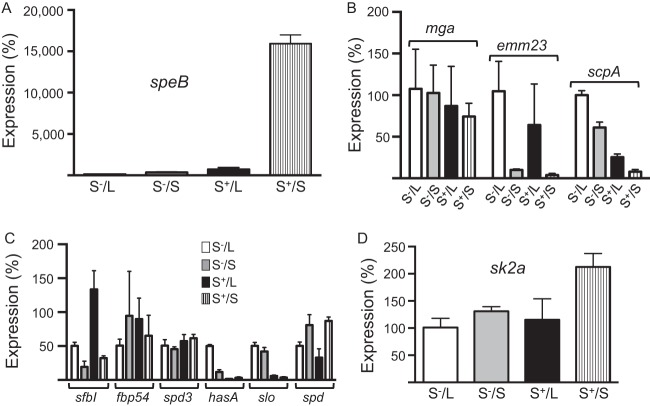
The first genome sequence of a group A Streptococcus pyogenes serotype M23 (emm23) strain (M23ND), isolated from an invasive human infection, has been completed. The genome of this opacity factor-negative (SOF(-)) strain is composed of a circular chromosome of 1,846,477 bp. Gene profiling showed that this strain contained six phage-encoded and 24 chromosomally inherited well-known virulence factors, as well as 11 pseudogenes. The bacterium has acquired four large prophage elements, ΦM23ND.1 to ΦM23ND.4, harboring genes encoding streptococcal superantigen (ssa), streptococcal pyrogenic exotoxins (speC, speH, and speI), and DNases (spd1 and spd3), with phage integrase genes being present at one flank of each phage insertion, suggesting that the phages were integrated by horizontal gene transfer. Comparative analyses revealed unique large-scale genomic rearrangements that result in genomic rearrangements that differ from those of previously sequenced GAS strains. These rearrangements resulted in an imbalanced genomic architecture and translocations of chromosomal virulence genes. The covS sensor in M23ND was identified as a pseudogene, resulting in the attenuation of speB function and increased expression of the genes for the chromosomal virulence factors multiple-gene activator (mga), M protein (emm23), C5a peptidase (scpA), fibronectin-binding proteins (sfbI and fbp54), streptolysin O (slo), hyaluronic acid capsule (hasA), streptokinase (ska), and DNases (spd and spd3), which were verified by PCR. These genes are responsible for facilitating host epithelial cell binding and and/or immune evasion, thus further contributing to the virulence of M23ND. In conclusion, strain M23ND has become highly pathogenic as the result of a combination of multiple genetic factors, particularly gene composition and mutations, prophage integrations, unique genomic rearrangements, and regulated expression of critical virulence factors.
Copyright © 2014, American Society for Microbiology. All Rights Reserved.
Figures
Similar articles
-
CovRS-Regulated Transcriptome Analysis of a Hypervirulent M23 Strain of Group A Streptococcus pyogenes Provides New Insights into Virulence Determinants.J Bacteriol. 2015 Oct;197(19):3191-205. doi: 10.1128/JB.00511-15. Epub 2015 Jul 27. J Bacteriol. 2015. PMID: 26216843 Free PMC article.
-
Genomic Characterization of a Pattern D Streptococcus pyogenes emm53 Isolate Reveals a Genetic Rationale for Invasive Skin Tropicity.J Bacteriol. 2016 May 27;198(12):1712-24. doi: 10.1128/JB.01019-15. Print 2016 Jun 15. J Bacteriol. 2016. PMID: 27044623 Free PMC article.
-
A response regulator that represses transcription of several virulence operons in the group A streptococcus.J Bacteriol. 1999 Jun;181(12):3649-57. doi: 10.1128/JB.181.12.3649-3657.1999. J Bacteriol. 1999. PMID: 10368137 Free PMC article.
-
The Bacteriophages of Streptococcus pyogenes.Microbiol Spectr. 2019 May;7(3):10.1128/microbiolspec.gpp3-0059-2018. doi: 10.1128/microbiolspec.GPP3-0059-2018. Microbiol Spectr. 2019. PMID: 31111820 Free PMC article. Review.
-
The DNases of pathogenic Lancefield streptococci.Microbiology (Reading). 2018 Mar;164(3):242-250. doi: 10.1099/mic.0.000612. Epub 2018 Jan 25. Microbiology (Reading). 2018. PMID: 29458565 Review.
Cited by
-
Streptococcus pyogenes Employs Strain-dependent Mechanisms of C3b Inactivation to Inhibit Phagocytosis and Killing of Bacteria.J Biol Chem. 2016 Apr 22;291(17):9181-9. doi: 10.1074/jbc.M115.704221. Epub 2016 Mar 4. J Biol Chem. 2016. PMID: 26945067 Free PMC article.
-
Molecular Epidemiology, Ecology, and Evolution of Group A Streptococci.Microbiol Spectr. 2018 Sep;6(5):10.1128/microbiolspec.cpp3-0009-2018. doi: 10.1128/microbiolspec.CPP3-0009-2018. Microbiol Spectr. 2018. PMID: 30191802 Free PMC article. Review.
-
Complete Genome Sequence of Streptococcus pyogenes emm28 Clinical Isolate M28PF1, Responsible for a Puerperal Fever.Genome Announc. 2015 Jul 16;3(4):e00750-15. doi: 10.1128/genomeA.00750-15. Genome Announc. 2015. PMID: 26184934 Free PMC article.
-
Variable region in streptococcal M-proteins provides stable binding with host fibrinogen for plasminogen-mediated bacterial invasion.J Biol Chem. 2017 Apr 21;292(16):6775-6785. doi: 10.1074/jbc.M116.768937. Epub 2017 Mar 9. J Biol Chem. 2017. PMID: 28280245 Free PMC article.
-
Phenotypic differentiation of Streptococcus pyogenes populations is induced by recombination-driven gene-specific sweeps.Sci Rep. 2016 Nov 8;6:36644. doi: 10.1038/srep36644. Sci Rep. 2016. PMID: 27821851 Free PMC article.
References
-
- Smoot JC, Barbian KD, Van Gompel JJ, Smoot LM, Chaussee MS, Sylva GL, Sturdevant DE, Ricklefs SM, Porcella SF, Parkins LD, Beres SB, Campbell DS, Smith TM, Zhang Q, Kapur V, Daly JA, Veasy LG, Musser JM. 2002. Genome sequence and comparative microarray analysis of serotype M18 group A Streptococcus strains associated with acute rheumatic fever outbreaks. Proc. Natl. Acad. Sci. U. S. A. 99:4668–4673. 10.1073/pnas.062526099. - DOI - PMC - PubMed
-
- Sanderson-Smith M, De Oliveira DM, Guglielmini J, McMillan DJ, Vu T, Holien JK, Henningham A, Steer AC, Bessen DE, Dale JB, Curtis N, Beall BW, Walker MJ, Parker MW, Carapetis JR, Van Melderen L, Sriprakash KS, Smeesters PR, the M Protein Study Group 5 May 2014. A systematic and functional classification of Streptococcus pyogenes that serves as a new tool for molecular typing and vaccine development. J. Infect. Dis. 10.1093/infdis/jiu260. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
