

Frustration in biomolecules
- PMID: 25225856
- PMCID: PMC4256721
- DOI: 10.1017/S0033583514000092
Frustration in biomolecules
Abstract
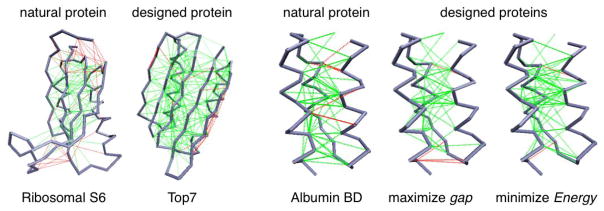
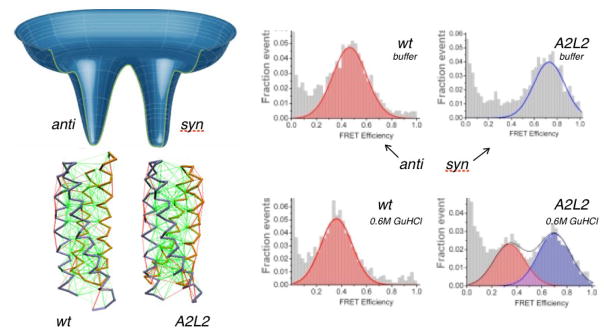
Biomolecules are the prime information processing elements of living matter. Most of these inanimate systems are polymers that compute their own structures and dynamics using as input seemingly random character strings of their sequence, following which they coalesce and perform integrated cellular functions. In large computational systems with finite interaction-codes, the appearance of conflicting goals is inevitable. Simple conflicting forces can lead to quite complex structures and behaviors, leading to the concept of frustration in condensed matter. We present here some basic ideas about frustration in biomolecules and how the frustration concept leads to a better appreciation of many aspects of the architecture of biomolecules, and especially how biomolecular structure connects to function by means of localized frustration. These ideas are simultaneously both seductively simple and perilously subtle to grasp completely. The energy landscape theory of protein folding provides a framework for quantifying frustration in large systems and has been implemented at many levels of description. We first review the notion of frustration from the areas of abstract logic and its uses in simple condensed matter systems. We discuss then how the frustration concept applies specifically to heteropolymers, testing folding landscape theory in computer simulations of protein models and in experimentally accessible systems. Studying the aspects of frustration averaged over many proteins provides ways to infer energy functions useful for reliable structure prediction. We discuss how frustration affects folding mechanisms. We review here how the biological functions of proteins are related to subtle local physical frustration effects and how frustration influences the appearance of metastable states, the nature of binding processes, catalysis and allosteric transitions. In this review, we also emphasize that frustration, far from being always a bad thing, is an essential feature of biomolecules that allows dynamics to be harnessed for function. In this way, we hope to illustrate how Frustration is a fundamental concept in molecular biology.
Figures




























References
-
- Adams Margaret Joan, et al. Structure of rhombohedral 2 zinc insulin crystals. Nature. 1969;224(5218):491–495.
-
- Anderson PW. Antiferromagnetism. Theory of Superexchange Interaction. Phys Rev. 1950;79(2):350–356. doi: 10.1103/PhysRev.79.350. url: http://link.aps.org/doi/10.1103/PhysRev.79.350. - DOI
-
- Anfinsen CB. Principles that govern the folding of protein chains. Science. 1973;181(4096):223–30. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
