

Streptolysin O and NAD-glycohydrolase prevent phagolysosome acidification and promote group A Streptococcus survival in macrophages
- PMID: 25227466
- PMCID: PMC4172074
- DOI: 10.1128/mBio.01690-14
Streptolysin O and NAD-glycohydrolase prevent phagolysosome acidification and promote group A Streptococcus survival in macrophages
Abstract
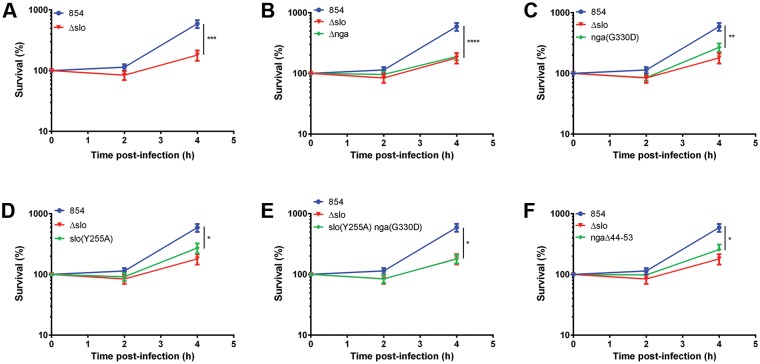
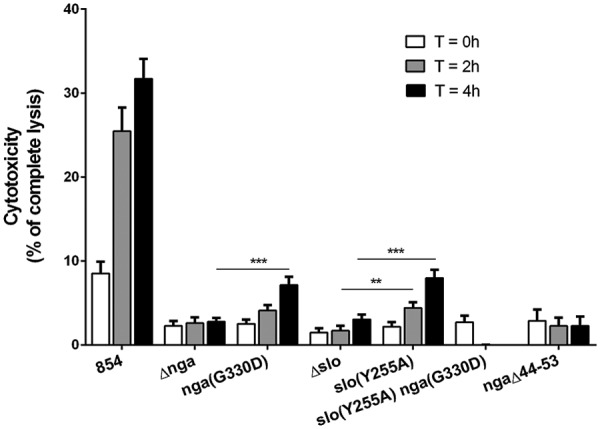
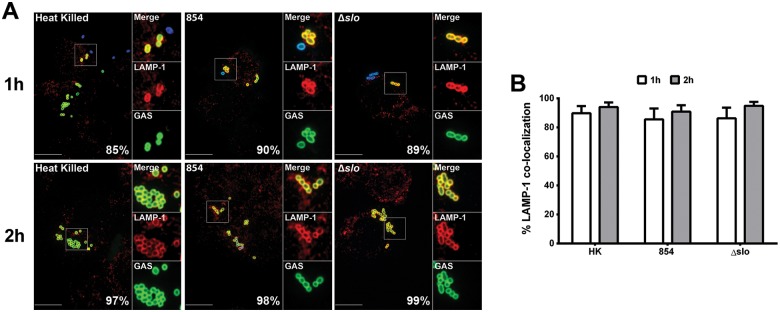
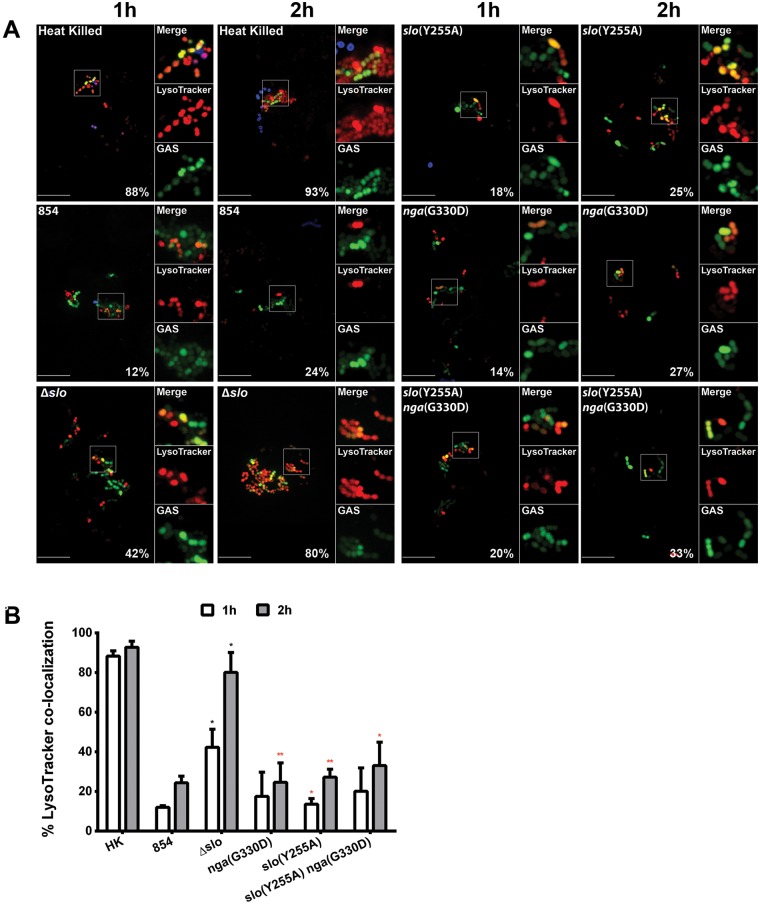
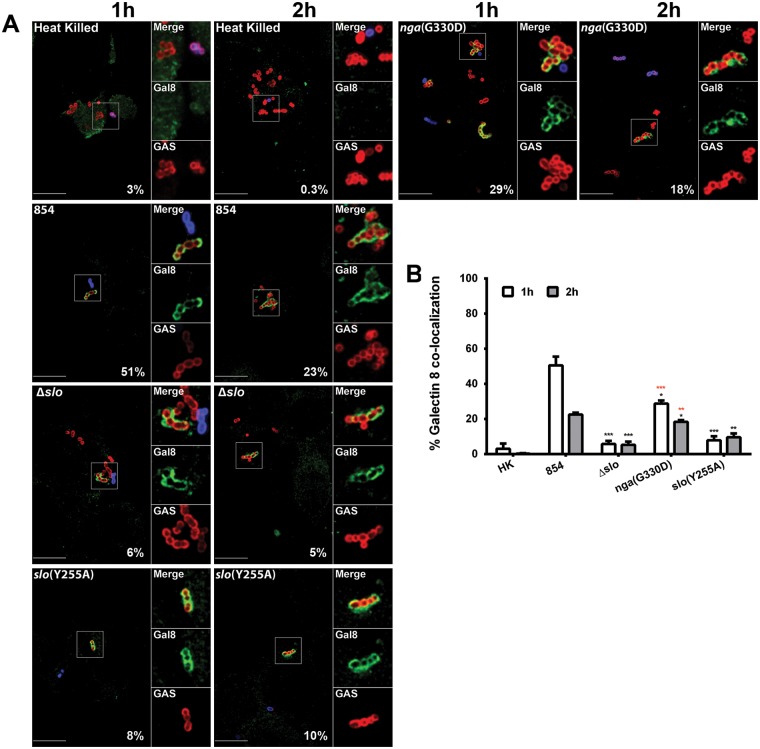
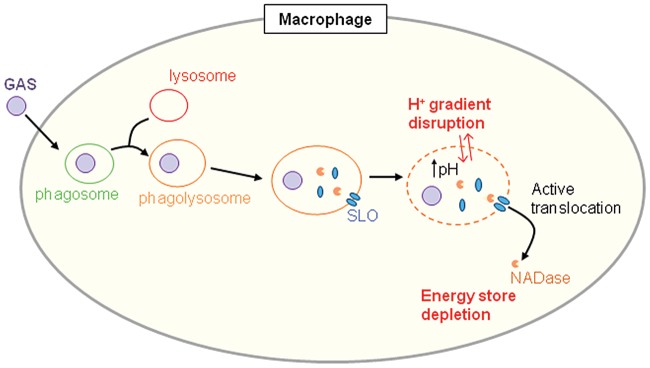
Group A Streptococcus (GAS, Streptococcus pyogenes) is an ongoing threat to human health as the agent of streptococcal pharyngitis, skin and soft tissue infections, and life-threatening conditions such as necrotizing fasciitis and streptococcal toxic shock syndrome. In animal models of infection, macrophages have been shown to contribute to host defense against GAS infection. However, as GAS can resist killing by macrophages in vitro and induce macrophage cell death, it has been suggested that GAS intracellular survival in macrophages may enable persistent infection. Using isogenic mutants, we now show that the GAS pore-forming toxin streptolysin O (SLO) and its cotoxin NAD-glycohydrolase (NADase) mediate GAS intracellular survival and cytotoxicity for macrophages. Unexpectedly, the two toxins did not inhibit fusion of GAS-containing phagosomes with lysosomes but rather prevented phagolysosome acidification. SLO served two essential functions, poration of the phagolysosomal membrane and translocation of NADase into the macrophage cytosol, both of which were necessary for maximal GAS intracellular survival. Whereas NADase delivery to epithelial cells is mediated by SLO secreted from GAS bound to the cell surface, in macrophages, the source of SLO and NADase is GAS contained within phagolysosomes. We found that transfer of NADase from the phagolysosome to the macrophage cytosol occurs not by simple diffusion through SLO pores but rather by a specific translocation mechanism that requires the N-terminal translocation domain of NADase. These results illuminate the mechanisms through which SLO and NADase enable GAS to defeat macrophage-mediated killing and provide new insight into the virulence of a major human pathogen.
Importance: Macrophages constitute an important element of the innate immune response to mucosal pathogens. They ingest and kill microbes by phagocytosis and secrete inflammatory cytokines to recruit and activate other effector cells. Group A Streptococcus (GAS, Streptococcus pyogenes), an important cause of pharyngitis and invasive infections, has been shown to resist killing by macrophages. We find that GAS resistance to macrophage killing depends on the GAS pore-forming toxin streptolysin O (SLO) and its cotoxin NAD-glycohydrolase (NADase). GAS bacteria are internalized by macrophage phagocytosis but resist killing by secreting SLO, which damages the phagolysosome membrane, prevents phagolysosome acidification, and translocates NADase from the phagolysosome into the macrophage cytosol. NADase augments SLO-mediated cytotoxicity by depleting cellular energy stores. These findings may explain the nearly universal production of SLO by GAS clinical isolates and the association of NADase with the global spread of a GAS clone implicated in invasive infections.
Copyright © 2014 Bastiat-Sempe et al.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
