

Structural insights into Bacillus thuringiensis Cry, Cyt and parasporin toxins
- PMID: 25229189
- PMCID: PMC4179158
- DOI: 10.3390/toxins6092732
Structural insights into Bacillus thuringiensis Cry, Cyt and parasporin toxins
Abstract
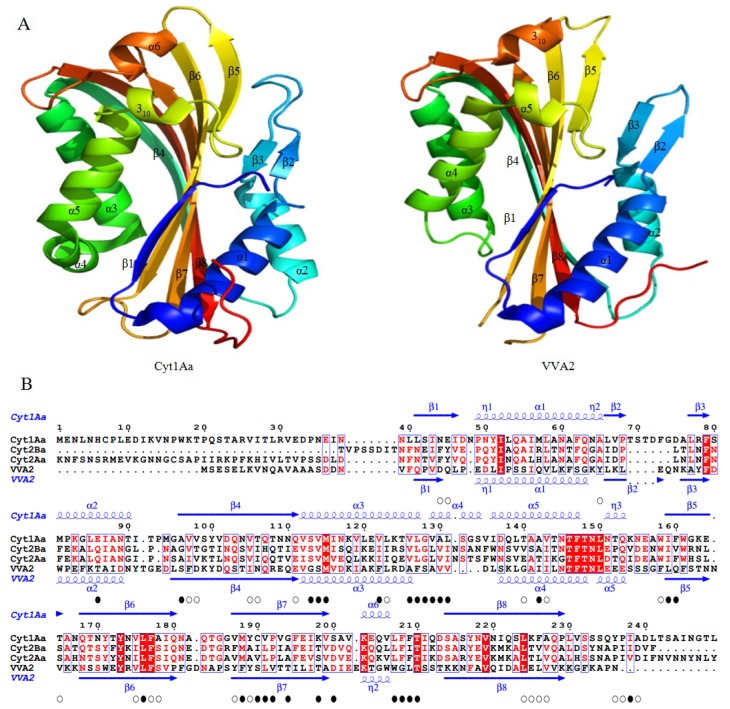
Since the first X-ray structure of Cry3Aa was revealed in 1991, numerous structures of B. thuringiensis toxins have been determined and published. In recent years, functional studies on the mode of action and resistance mechanism have been proposed, which notably promoted the developments of biological insecticides and insect-resistant transgenic crops. With the exploration of known pore-forming toxins (PFTs) structures, similarities between PFTs and B. thuringiensis toxins have provided great insights into receptor binding interactions and conformational changes from water-soluble to membrane pore-forming state of B. thuringiensis toxins. This review mainly focuses on the latest discoveries of the toxin working mechanism, with the emphasis on structural related progress. Based on the structural features, B. thuringiensis Cry, Cyt and parasporin toxins could be divided into three categories: three-domain type α-PFTs, Cyt toxin type β-PFTs and aerolysin type β-PFTs. Structures from each group are elucidated and discussed in relation to the latest data, respectively.
Figures






Similar articles
-
The pesticidal Cry6Aa toxin from Bacillus thuringiensis is structurally similar to HlyE-family alpha pore-forming toxins.BMC Biol. 2016 Aug 30;14(1):71. doi: 10.1186/s12915-016-0295-9. BMC Biol. 2016. PMID: 27576487 Free PMC article.
-
Mode of action of Bacillus thuringiensis Cry and Cyt toxins and their potential for insect control.Toxicon. 2007 Mar 15;49(4):423-35. doi: 10.1016/j.toxicon.2006.11.022. Epub 2006 Nov 30. Toxicon. 2007. PMID: 17198720 Free PMC article. Review.
-
Pore formation by Cry toxins.Adv Exp Med Biol. 2010;677:127-42. doi: 10.1007/978-1-4419-6327-7_11. Adv Exp Med Biol. 2010. PMID: 20687486 Review.
-
Potential Prepore Trimer Formation by the Bacillus thuringiensis Mosquito-specific Toxin: MOLECULAR INSIGHTS INTO A CRITICAL PREREQUISITE OF MEMBRANE-BOUND MONOMERS.J Biol Chem. 2015 Aug 21;290(34):20793-20803. doi: 10.1074/jbc.M114.627554. Epub 2015 Jun 25. J Biol Chem. 2015. PMID: 26112409 Free PMC article.
-
Specific binding between Bacillus thuringiensis Cry9Aa and Vip3Aa toxins synergizes their toxicity against Asiatic rice borer (Chilo suppressalis).J Biol Chem. 2018 Jul 20;293(29):11447-11458. doi: 10.1074/jbc.RA118.003490. Epub 2018 Jun 1. J Biol Chem. 2018. PMID: 29858245 Free PMC article.
Cited by
-
A Review of the Functional Annotations of Important Genes in the AHPND-Causing pVA1 Plasmid.Microorganisms. 2020 Jul 3;8(7):996. doi: 10.3390/microorganisms8070996. Microorganisms. 2020. PMID: 32635298 Free PMC article. Review.
-
The opportunistic marine pathogen Vibrio parahaemolyticus becomes virulent by acquiring a plasmid that expresses a deadly toxin.Proc Natl Acad Sci U S A. 2015 Aug 25;112(34):10798-803. doi: 10.1073/pnas.1503129112. Epub 2015 Aug 10. Proc Natl Acad Sci U S A. 2015. PMID: 26261348 Free PMC article.
-
Differentiation of Bacillus cereus and Bacillus thuringiensis Using Genome-Guided MALDI-TOF MS Based on Variations in Ribosomal Proteins.Microorganisms. 2022 Apr 27;10(5):918. doi: 10.3390/microorganisms10050918. Microorganisms. 2022. PMID: 35630362 Free PMC article.
-
Structural and biophysical characterization of Bacillus thuringiensis insecticidal proteins Cry34Ab1 and Cry35Ab1.PLoS One. 2014 Nov 12;9(11):e112555. doi: 10.1371/journal.pone.0112555. eCollection 2014. PLoS One. 2014. PMID: 25390338 Free PMC article.
-
Functional characterization of Vip3Ab1 and Vip3Bc1: Two novel insecticidal proteins with differential activity against lepidopteran pests.Sci Rep. 2017 Sep 11;7(1):11112. doi: 10.1038/s41598-017-11702-2. Sci Rep. 2017. PMID: 28894249 Free PMC article.
References
-
- Nester E.W., Thomashow L.S., Metz M. 100 Years of Bacillus thuringiensis: A Critical Scientific Assessment. American Society for Microbiology (ASM); Washington, DC, USA: 2002. - PubMed
-
- De Barjac H., Bonnefoi A. Essai de classification biochimique et sérologique de 24 souches de Bacillus du type B. thuringiensis. Entomophaga. 1962;7:5–31. doi: 10.1007/BF02375988. (In French) - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
