

Systematic dissection of the mechanisms underlying progesterone receptor downregulation in endometrial cancer
- PMID: 25229191
- PMCID: PMC4259437
- DOI: 10.18632/oncotarget.2392
Systematic dissection of the mechanisms underlying progesterone receptor downregulation in endometrial cancer
Abstract
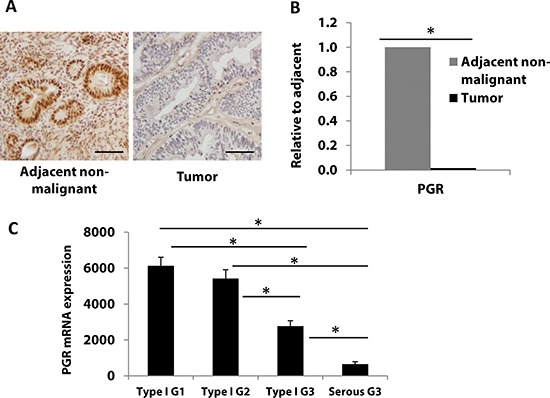
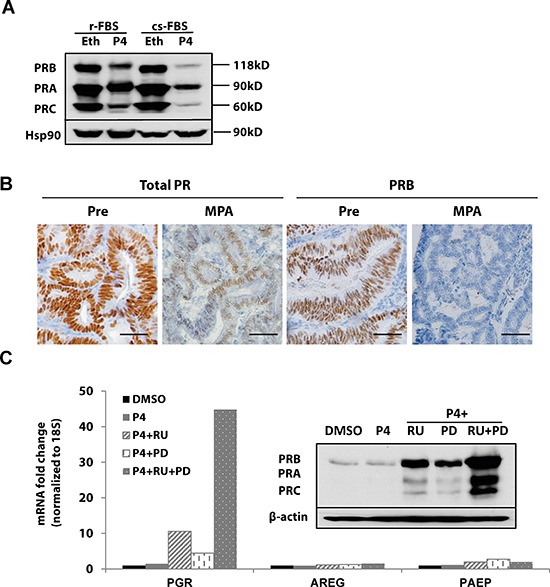
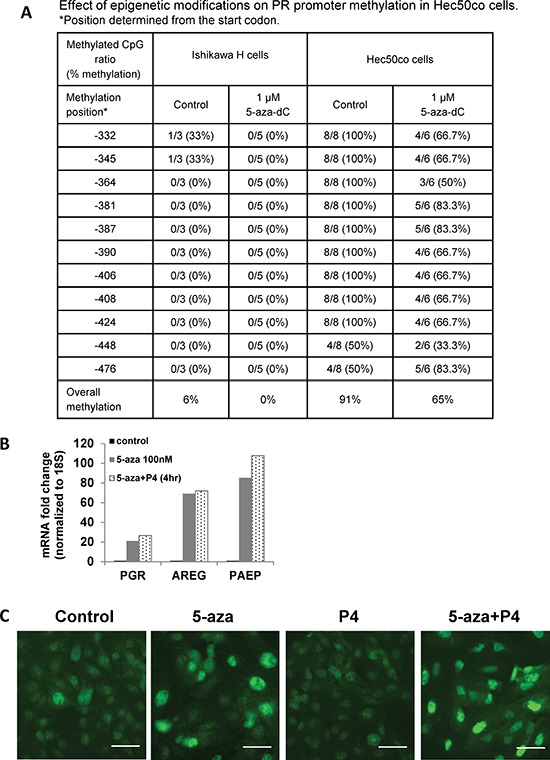
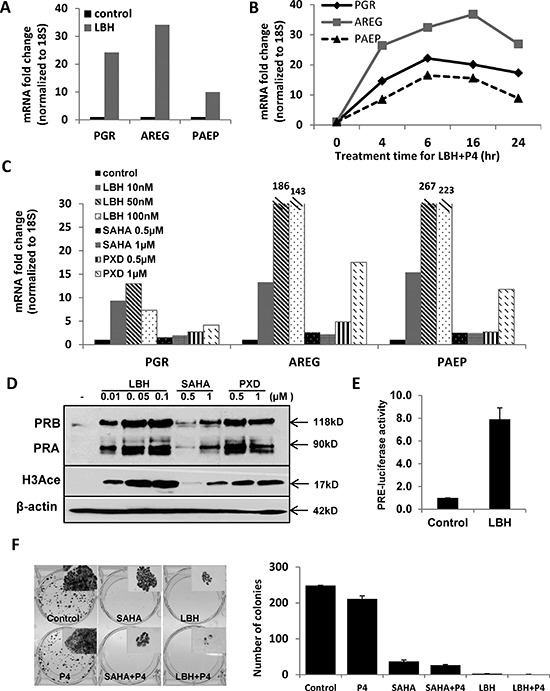
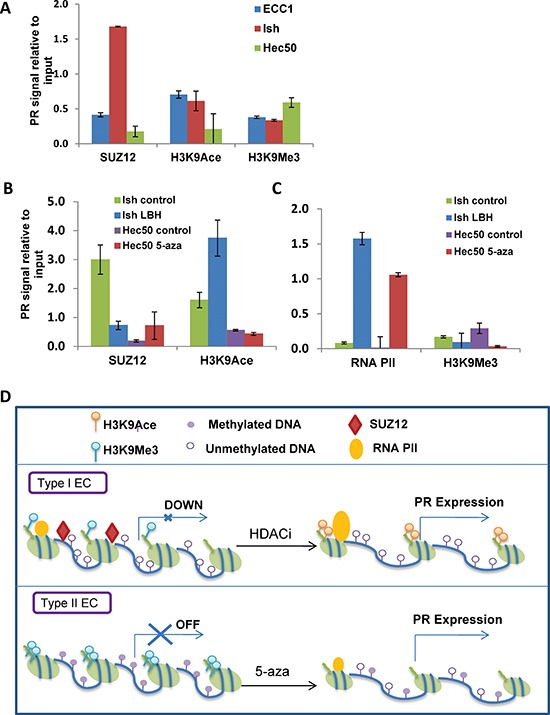
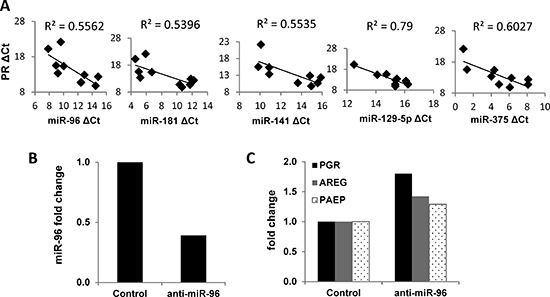
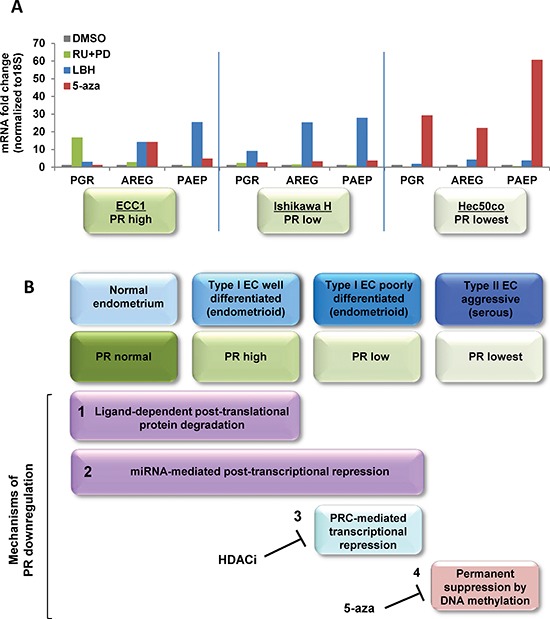
Progesterone, acting through its receptor, PR (progesterone receptor), is the natural inhibitor of uterine endometrial carcinogenesis by inducing differentiation. PR is downregulated in more advanced cases of endometrial cancer, thereby limiting the effectiveness of hormonal therapy. Our objective was to understand and reverse the mechanisms underlying loss of PR expression in order to improve therapeutic outcomes. Using endometrial cancer cell lines and data from The Cancer Genome Atlas, our findings demonstrate that PR expression is downregulated at four distinct levels. In well-differentiated cancers, ligand-induced receptor activation and downregulation are intact. miRNAs mediate fine tuning of PR levels. As differentiation is lost, PR silencing is primarily at the epigenetic level. Initially, recruitment of the polycomb repressor complex 2 to the PR promoter suppresses transcription. Subsequently, DNA methylation prevents PR expression. Appropriate epigenetic modulators reverse these mechanisms. These data provide a rationale for combining epigenetic modulators with progestins as a therapeutic strategy for endometrial cancer.
Significance: Traditional hormonal therapy for women with endometrial cancer can be molecularly enhanced by combining progestins with epigenetic modulators, thereby increasing progesterone receptor expression and significantly improving treatment efficacy.
Figures
References
-
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11–30. - PubMed
-
- Singh M, Zaino RJ, Filiaci VJ, Leslie KK. Relationship of estrogen and progesterone receptors to clinical outcome in metastatic endometrial carcinoma: a Gynecologic Oncology Group Study. Gynecol Oncol. 2007;106(2):325–333. - PubMed
-
- Dahmoun M, Backstrom T, Boman K, Cajander S. Apoptosis, proliferation, and hormone receptors in endometrial carcinoma: results depending on methods of analysis. Int J Oncol. 2003;22(1):115–122. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
