

BNIP3 interacting with LC3 triggers excessive mitophagy in delayed neuronal death in stroke
- PMID: 25230377
- PMCID: PMC6492992
- DOI: 10.1111/cns.12325
BNIP3 interacting with LC3 triggers excessive mitophagy in delayed neuronal death in stroke
Abstract
Introduction: A basal level of mitophagy is essential in mitochondrial quality control in physiological conditions, while excessive mitophagy contributes to cell death in a number of diseases including ischemic stroke. Signals regulating this process remain unknown. BNIP3, a pro-apoptotic BH3-only protein, has been implicated as a regulator of mitophagy.
Aims: Both in vivo and in vitro models of stroke, as well as BNIP3 wild-type and knock out mice were used in this study.
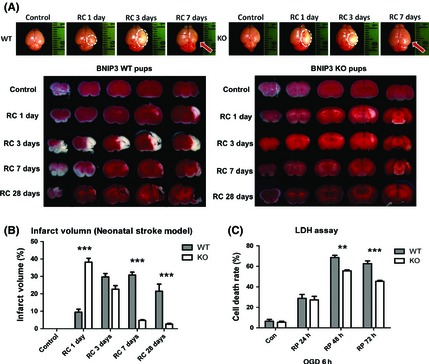
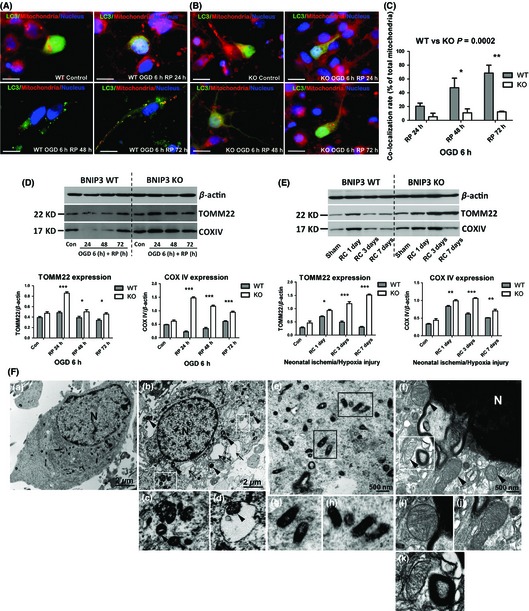
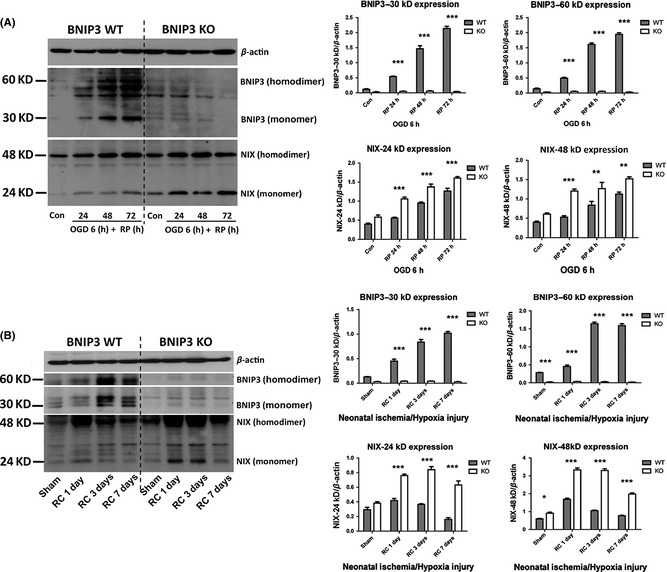
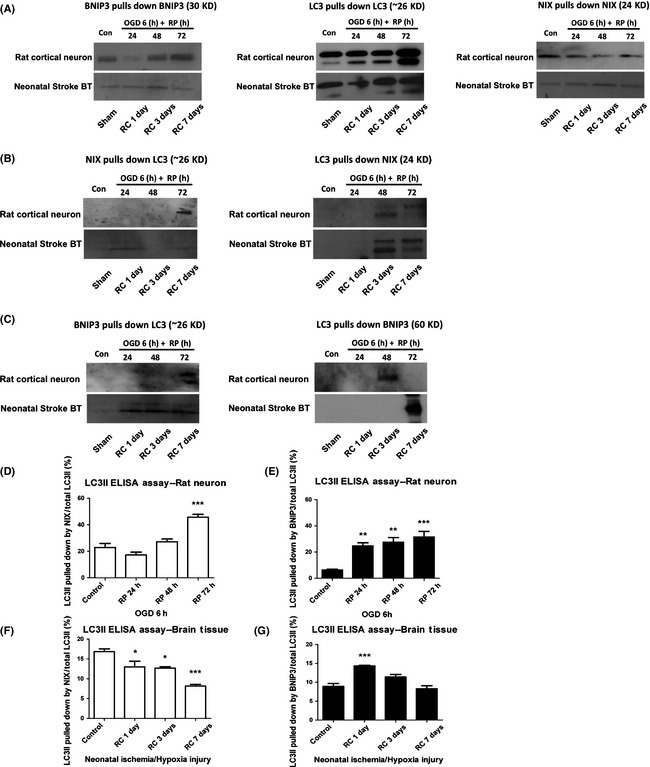
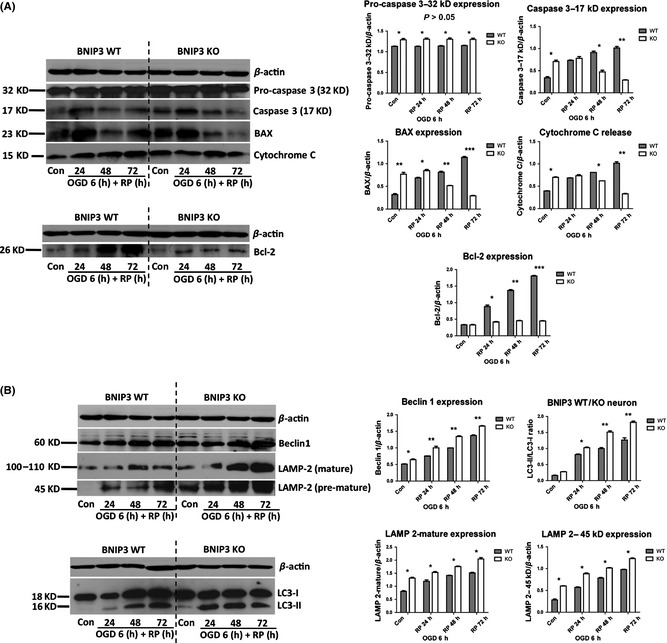
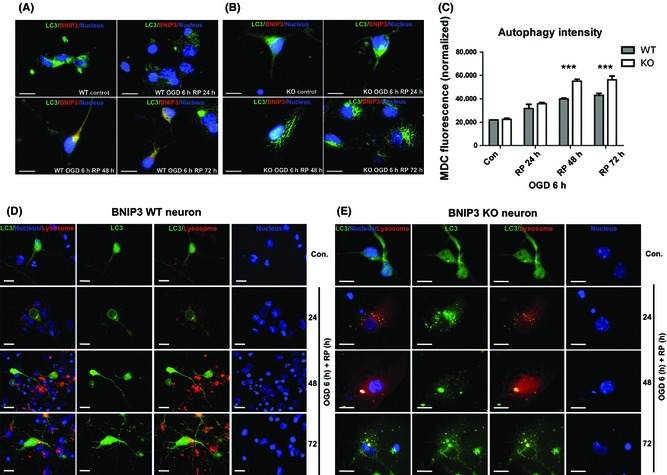
Results: We show that BNIP3 and its homologue BNIP3L (NIX) are highly expressed in a "delayed" manner and contribute to delayed neuronal loss following stroke. Deficiency in BNIP3 significantly decreases both neuronal mitophagy and apoptosis but increases nonselective autophagy following ischemic/hypoxic insults. The mitochondria-localized BNIP3 interacts with the autophagosome-localized LC3, suggesting that BNIP3, similar to NIX, functions as a LC3-binding receptor on mitochondria. Although NIX expression is upregulated when BNIP3 is silenced, up-regulation of NIX cannot functionally compensate for the loss of BNIP3 in activating excessive mitophagy.
Conclusions: NIX primarily regulates basal level of mitophagy in physiological conditions, whereas BNIP3 exclusively activates excessive mitophagy leading to cell death.
Keywords: BNIP3; LC3; Mitophagy; NIX; Neonatal stroke.
© 2014 John Wiley & Sons Ltd.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Bcl-2/E1B-19KD-Interacting Protein 3/Light Chain 3 Interaction Induces Mitophagy in Spinal Cord Injury in Rats Both In Vivo and In Vitro.J Neurotrauma. 2018 Sep 15;35(18):2183-2194. doi: 10.1089/neu.2017.5280. Epub 2018 Jun 7. J Neurotrauma. 2018. PMID: 29566574
-
Role of BNIP3 and NIX in cell death, autophagy, and mitophagy.Cell Death Differ. 2009 Jul;16(7):939-46. doi: 10.1038/cdd.2009.16. Epub 2009 Feb 20. Cell Death Differ. 2009. PMID: 19229244 Free PMC article. Review.
-
NIX Mediates Mitophagy in Spinal Cord Injury in Rats by Interacting with LC3.Cell Mol Neurobiol. 2022 Aug;42(6):1983-1994. doi: 10.1007/s10571-021-01082-7. Epub 2021 Mar 29. Cell Mol Neurobiol. 2022. PMID: 33779883 Free PMC article.
-
Induction of neuronal mitophagy in acute spinal cord injury in rats.Neurotox Res. 2013 Nov;24(4):512-22. doi: 10.1007/s12640-013-9397-0. Epub 2013 Apr 30. Neurotox Res. 2013. PMID: 23637053
-
Coordinating BNIP3/NIX-mediated mitophagy in space and time.Biochem Soc Trans. 2024 Oct 30;52(5):1969-1979. doi: 10.1042/BST20221364. Biochem Soc Trans. 2024. PMID: 39377319 Free PMC article. Review.
Cited by
-
Mitophagy-Mediated Tumor Dormancy Protects Cancer Cells from Chemotherapy.Biomedicines. 2024 Jan 28;12(2):305. doi: 10.3390/biomedicines12020305. Biomedicines. 2024. PMID: 38397907 Free PMC article.
-
Knockdown of BNIP3L or SQSTM1 alters cellular response to mitochondria target drugs.Autophagy. 2019 May;15(5):900-907. doi: 10.1080/15548627.2018.1558002. Epub 2019 Jan 9. Autophagy. 2019. PMID: 30563411 Free PMC article.
-
BNIP3L/NIX-mediated mitophagy: molecular mechanisms and implications for human disease.Cell Death Dis. 2021 Dec 20;13(1):14. doi: 10.1038/s41419-021-04469-y. Cell Death Dis. 2021. PMID: 34930907 Free PMC article. Review.
-
HIF-1α Activation Promotes Luteolysis by Enhancing ROS Levels in the Corpus Luteum of Pseudopregnant Rats.Oxid Med Cell Longev. 2021 Sep 1;2021:1764929. doi: 10.1155/2021/1764929. eCollection 2021. Oxid Med Cell Longev. 2021. PMID: 34512862 Free PMC article.
-
The BDNF mimetic R-13 attenuates TBI pathogenesis using TrkB-related pathways and bioenergetics.Biochim Biophys Acta Mol Basis Dis. 2023 Oct;1869(7):166781. doi: 10.1016/j.bbadis.2023.166781. Epub 2023 Jun 5. Biochim Biophys Acta Mol Basis Dis. 2023. PMID: 37286142 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
