

Antithrombin up-regulates AMP-activated protein kinase signalling during myocardial ischaemia/reperfusion injury
- PMID: 25230600
- PMCID: PMC4308562
- DOI: 10.1160/TH14-04-0360
Antithrombin up-regulates AMP-activated protein kinase signalling during myocardial ischaemia/reperfusion injury
Abstract
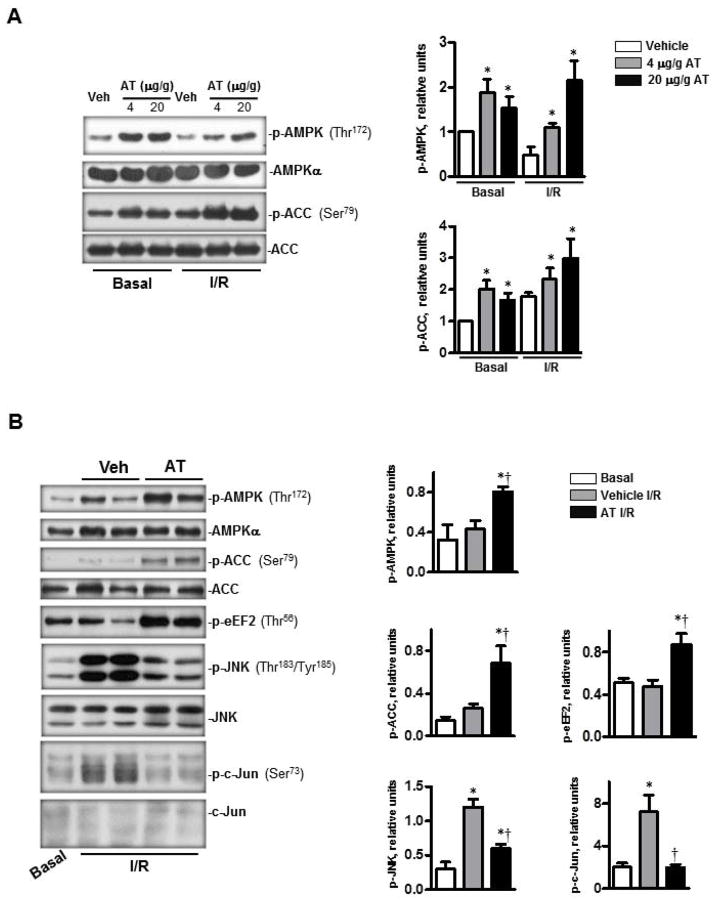
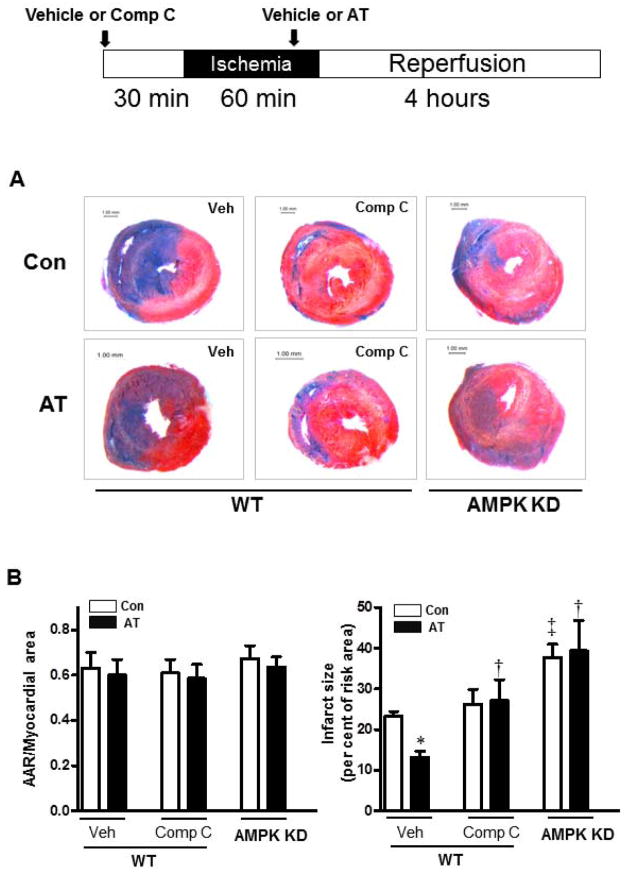
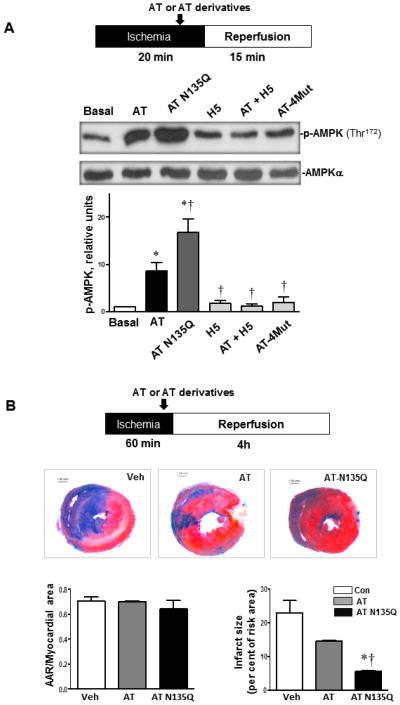
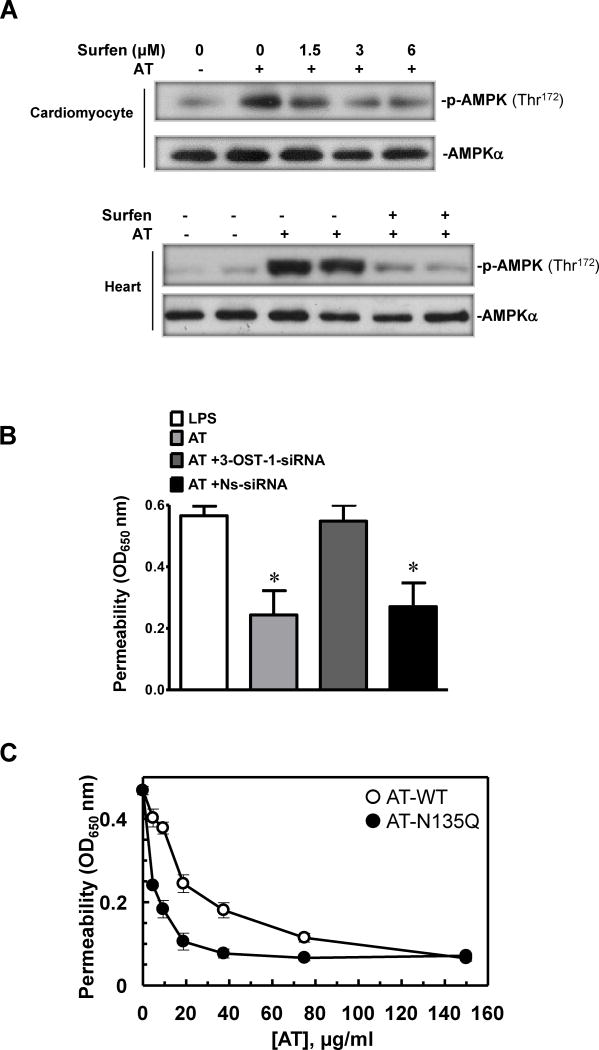
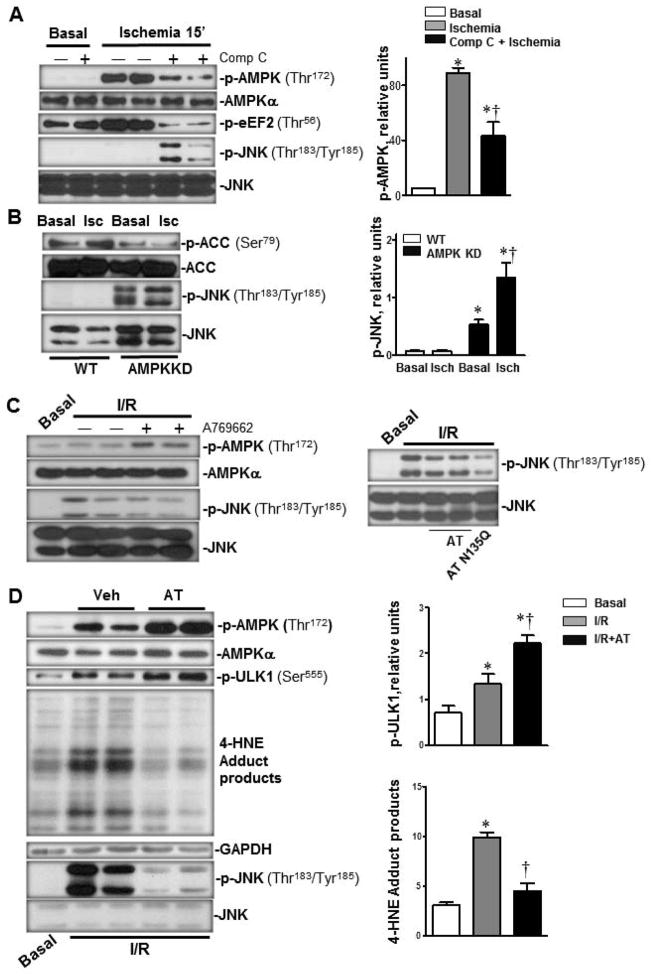
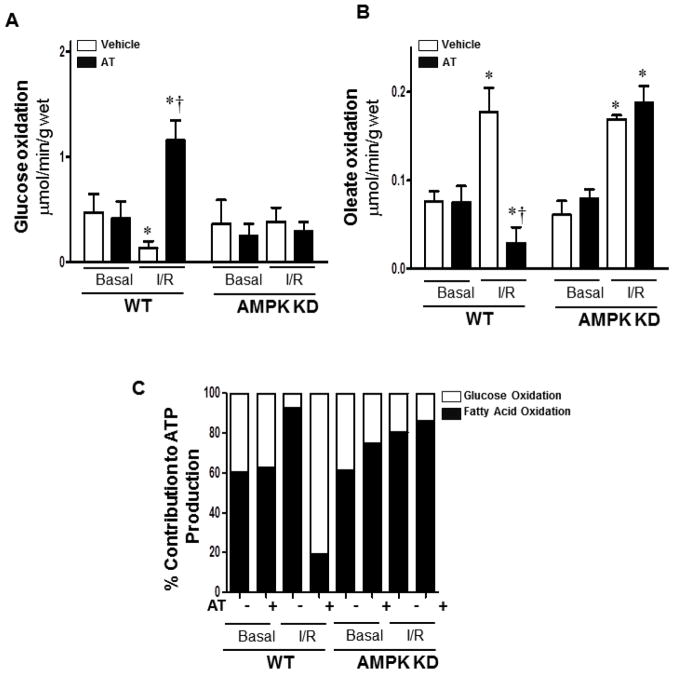
Antithrombin (AT) is a protein of the serpin superfamily involved in regulation of the proteolytic activity of the serine proteases of the coagulation system. AT is known to exhibit anti-inflammatory and cardioprotective properties when it binds to heparan sulfate proteoglycans (HSPGs) on vascular cells. AMP-activated protein kinase (AMPK) plays an important cardioprotective role during myocardial ischaemia and reperfusion (I/R). To determine whether the cardioprotective signaling function of AT is mediated through the AMPK pathway, we evaluated the cardioprotective activities of wild-type AT and its two derivatives, one having high affinity and the other no affinity for heparin, in an acute I/R injury model in C57BL/6J mice in which the left anterior descending coronary artery was occluded. The serpin derivatives were given 5 minutes before reperfusion. The results showed that AT-WT can activate AMPK in both in vivo and ex vivo conditions. Blocking AMPK activity abolished the cardioprotective function of AT against I/R injury. The AT derivative having high affinity for heparin was more effective in activating AMPK and in limiting infraction, but the derivative lacking affinity for heparin was inactive in eliciting AMPK-dependent cardioprotective activity. Activation of AMPK by AT inhibited the inflammatory c-Jun N-terminal protein kinase (JNK) pathway during I/R. Further studies revealed that the AMPK activity induced by AT also modulates cardiac substrate metabolism by increasing glucose oxidation but inhibiting fatty acid oxidation during I/R. These results suggest that AT binds to HSPGs on heart tissues to invoke a cardioprotective function by triggering cardiac AMPK activation, thereby attenuating JNK inflammatory signalling pathways and modulating substrate metabolism during I/R.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
References
-
- Ferdinandy P, Schulz R, Baxter GF. Interaction of cardiovascular risk factors with myocardial ischemia/reperfusion injury, preconditioning, and postconditioning. Pharmacol Rev. 2007;59:418–458. - PubMed
-
- Hardie DG. Minireview: the AMP-activated protein kinase cascade: the key sensor of cellular energy status. Endocrinology. 2003;144:5179–5183. - PubMed
-
- Dolinsky VW, Dyck JR. Role of AMP-activated protein kinase in healthy and diseased hearts. Am J Physiol Heart Circ Physiol. 2006;291:H2557–2569. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
