

Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer's disease
- PMID: 25231068
- PMCID: PMC4207354
- DOI: 10.1186/s40478-014-0135-5
Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer's disease
Abstract
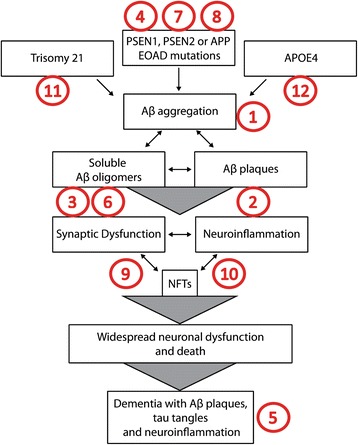
The amyloid hypothesis has driven drug development strategies for Alzheimer's disease for over 20 years. We review why accumulation of amyloid-beta (Aβ) oligomers is generally considered causal for synaptic loss and neurodegeneration in AD. We elaborate on and update arguments for and against the amyloid hypothesis with new data and interpretations, and consider why the amyloid hypothesis may be failing therapeutically. We note several unresolved issues in the field including the presence of Aβ deposition in cognitively normal individuals, the weak correlation between plaque load and cognition, questions regarding the biochemical nature, presence and role of Aβ oligomeric assemblies in vivo, the bias of pre-clinical AD models toward the amyloid hypothesis and the poorly explained pathological heterogeneity and comorbidities associated with AD. We also illustrate how extensive data cited in support of the amyloid hypothesis, including genetic links to disease, can be interpreted independently of a role for Aβ in AD. We conclude it is essential to expand our view of pathogenesis beyond Aβ and tau pathology and suggest several future directions for AD research, which we argue will be critical to understanding AD pathogenesis.
Figures


References
-
- Glenner GG, Wong CW. Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120:885–890. - PubMed
-
- Masters CL. Etiology and pathogenesis of Alzheimer's disease. Pathology. 1984;16:233–234. - PubMed
-
- Hardy J, Mayer J. The amyloid cascade hypothesis has misled the pharmaceutical industry. Biochem Soc Trans. 2011;39:920–923. - PubMed
-
- Masters CL, Gajdusek DC, Gibbs CJ., Jr The familial occurrence of Creutzfeldt-Jakob disease and Alzheimer's disease. Brain. 1981;104:535–558. - PubMed
-
- Prusiner SB, McKinley MP, Bowman KA, Bolton DC, Bendheim PE, Groth DF, Glenner GG. Scrapie prions aggregate to form amyloid-like birefringent rods. Cell. 1983;35:349–358. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
