

Analysis of the evolution and structure of a complex intrahost viral population in chronic hepatitis C virus mapped by ultradeep pyrosequencing
- PMID: 25231312
- PMCID: PMC4248971
- DOI: 10.1128/JVI.01732-14
Analysis of the evolution and structure of a complex intrahost viral population in chronic hepatitis C virus mapped by ultradeep pyrosequencing
Abstract
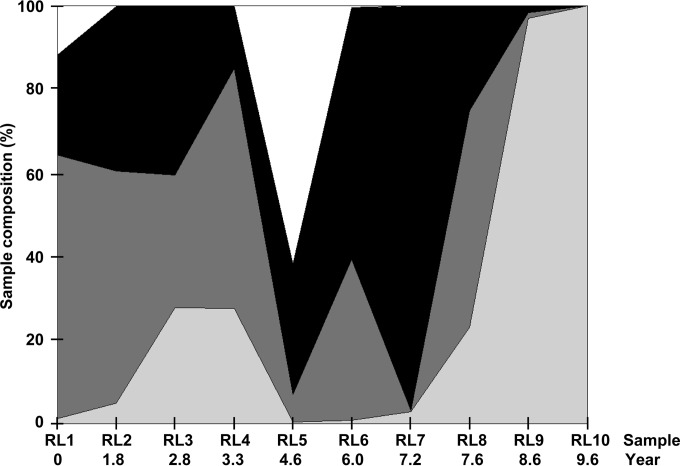
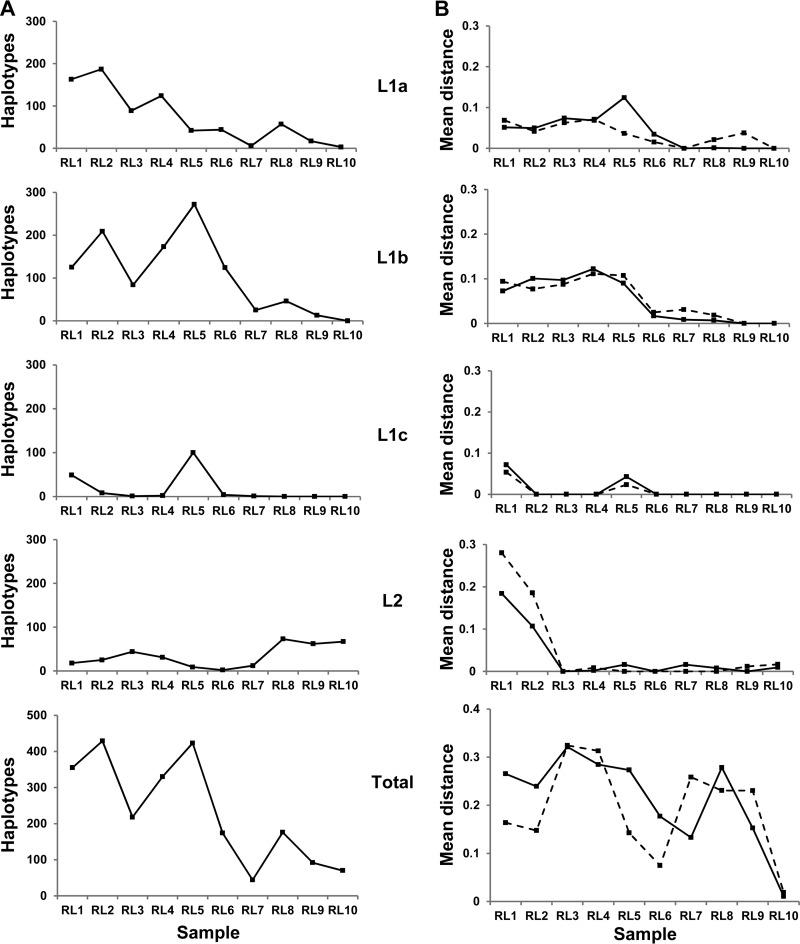
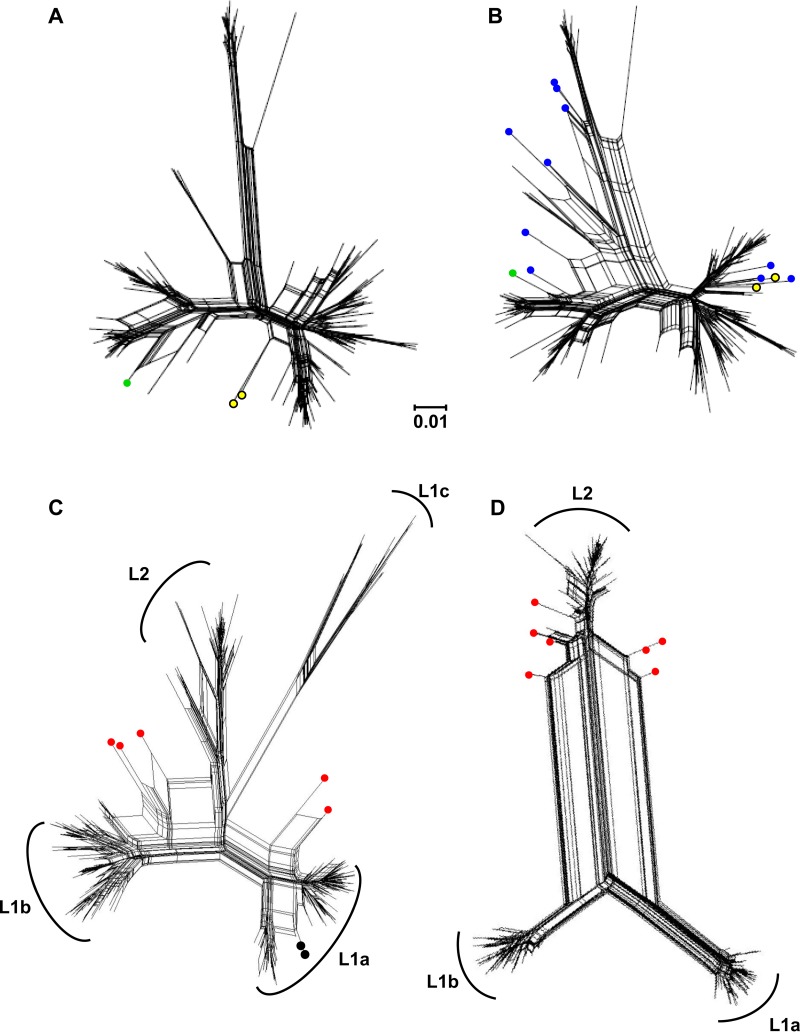
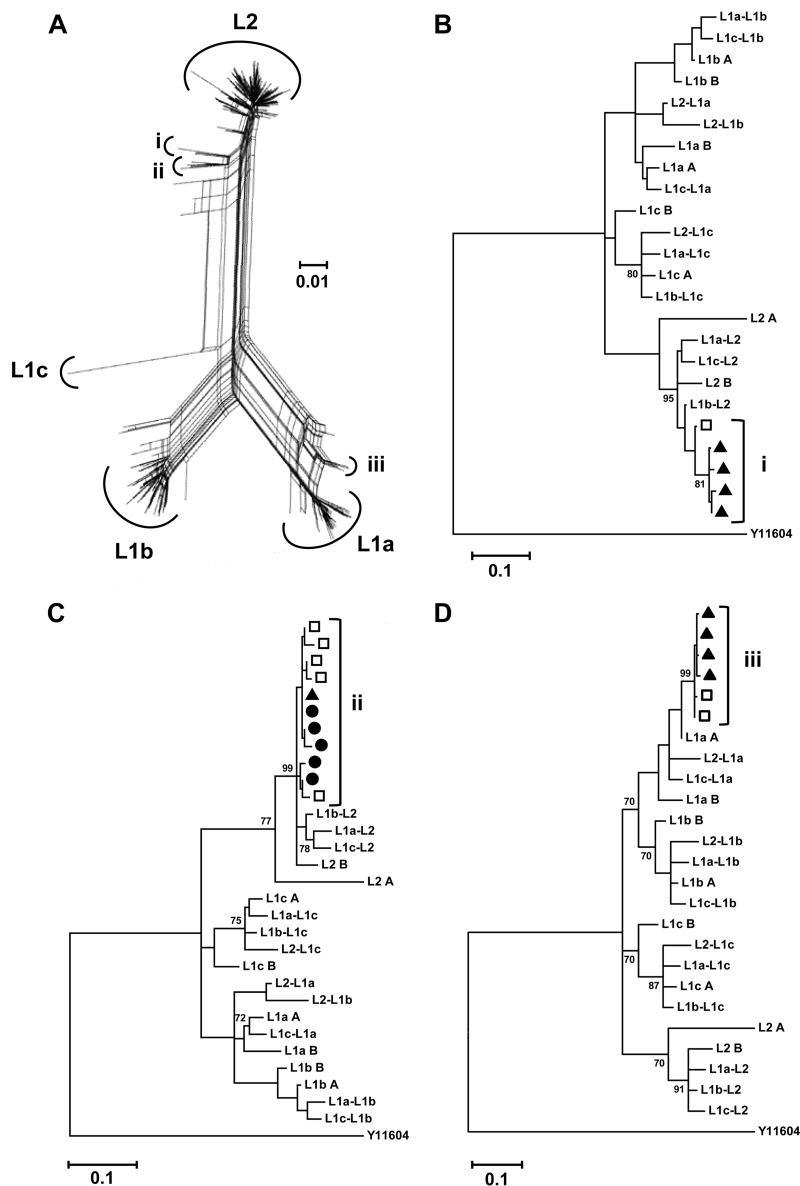
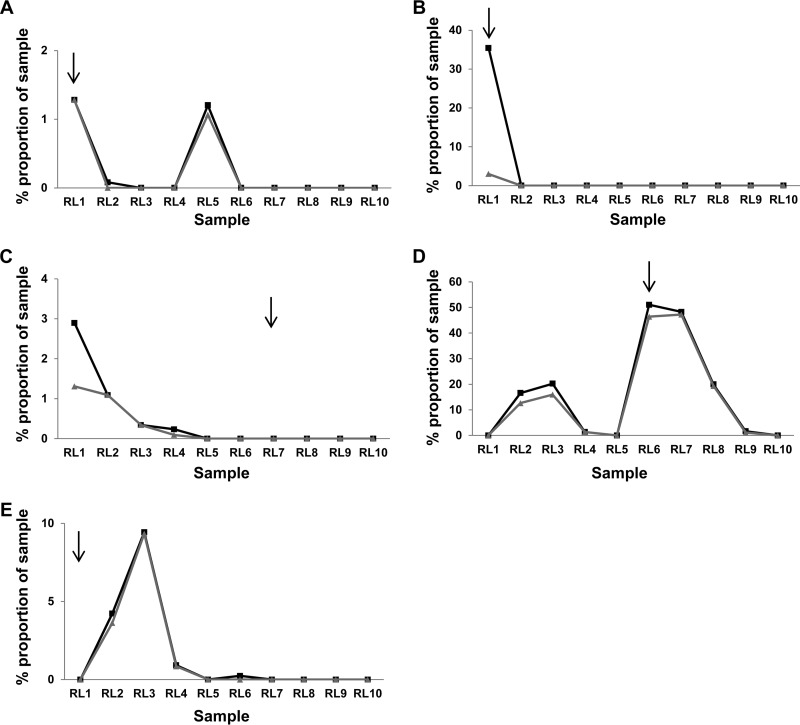
Hepatitis C virus (HCV) causes chronic infection in up to 50% to 80% of infected individuals. Hypervariable region 1 (HVR1) variability is frequently studied to gain an insight into the mechanisms of HCV adaptation during chronic infection, but the changes to and persistence of HCV subpopulations during intrahost evolution are poorly understood. In this study, we used ultradeep pyrosequencing (UDPS) to map the viral heterogeneity of a single patient over 9.6 years of chronic HCV genotype 4a infection. Informed error correction of the raw UDPS data was performed using a temporally matched clonal data set. The resultant data set reported the detection of low-frequency recombinants throughout the study period, implying that recombination is an active mechanism through which HCV can explore novel sequence space. The data indicate that polyvirus infection of hepatocytes has occurred but that the fitness quotients of recombinant daughter virions are too low for the daughter virions to compete against the parental genomes. The subpopulations of parental genomes contributing to the recombination events highlighted a dynamic virome where subpopulations of variants are in competition. In addition, we provide direct evidence that demonstrates the growth of subdominant populations to dominance in the absence of a detectable humoral response.
Importance: Analysis of ultradeep pyrosequencing data sets derived from virus amplicons frequently relies on software tools that are not optimized for amplicon analysis, assume random incorporation of sequencing errors, and are focused on achieving higher specificity at the expense of sensitivity. Such analysis is further complicated by the presence of hypervariable regions. In this study, we made use of a temporally matched reference sequence data set to inform error correction algorithms. Using this methodology, we were able to (i) detect multiple instances of hepatitis C virus intrasubtype recombination at the E1/E2 junction (a phenomenon rarely reported in the literature) and (ii) interrogate the longitudinal quasispecies complexity of the virome. Parallel to the UDPS, isolation of IgG-bound virions was found to coincide with the collapse of specific viral subpopulations.
Copyright © 2014, American Society for Microbiology. All Rights Reserved.
Figures
References
-
- Penin F, Combet C, Germanidis G, Frainais PO, Deleage G, Pawlotsky JM. 2001. Conservation of the conformation and positive charges of hepatitis C virus E2 envelope glycoprotein hypervariable region 1 points to a role in cell attachment. J. Virol. 75:5703–5710. 10.1128/JVI.75.12.5703-5710.2001. - DOI - PMC - PubMed
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
