

Implication of the cause of differences in 3D structures of proteins with high sequence identity based on analyses of amino acid sequences and 3D structures
- PMID: 25231773
- PMCID: PMC4180342
- DOI: 10.1186/1756-0500-7-654
Implication of the cause of differences in 3D structures of proteins with high sequence identity based on analyses of amino acid sequences and 3D structures
Abstract
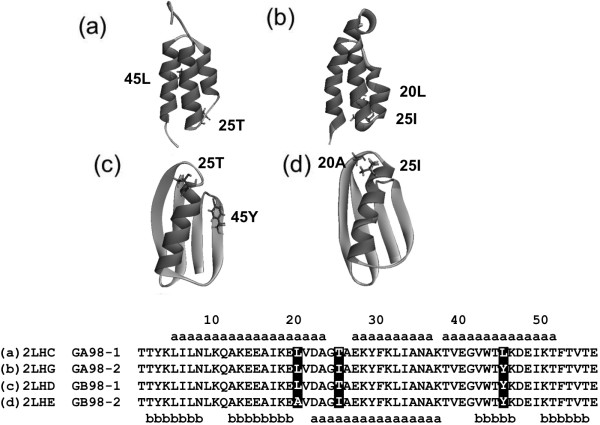
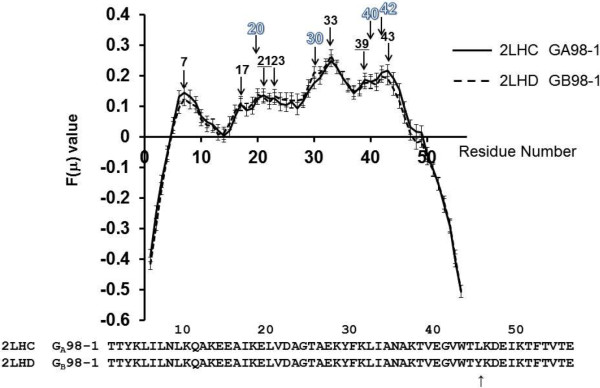
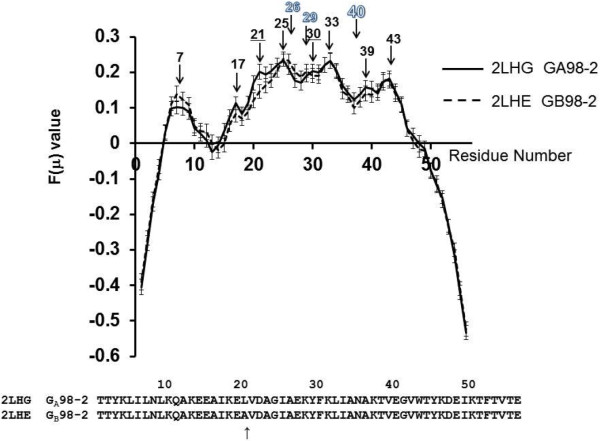
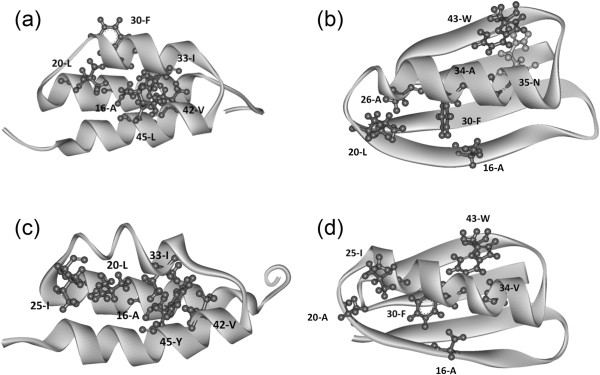
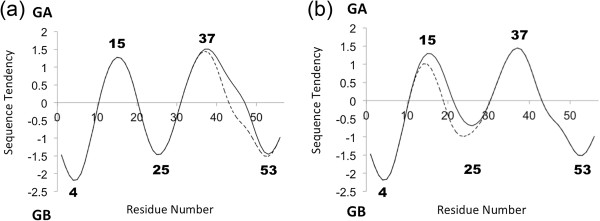
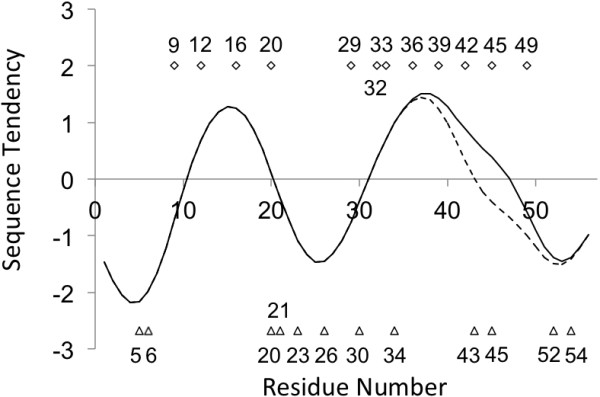
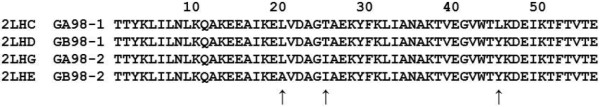
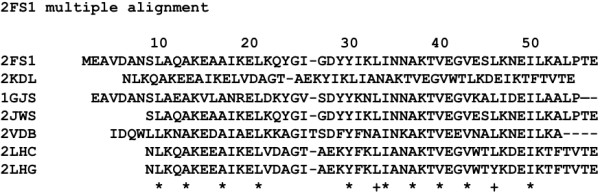
Background: Proteins that share a high sequence homology while exhibiting drastically different 3D structures are investigated in this study. Recently, artificial proteins related to the sequences of the GA and IgG binding GB domains of human serum albumin have been designed. These artificial proteins, referred to as GA and GB, share 98% amino acid sequence identity but exhibit different 3D structures, namely, a 3α bundle versus a 4β + α structure. Discriminating between their 3D structures based on their amino acid sequences is a very difficult problem. In the present work, in addition to using bioinformatics techniques, an analysis based on inter-residue average distance statistics is used to address this problem.
Results: It was hard to distinguish which structure a given sequence would take only with the results of ordinary analyses like BLAST and conservation analyses. However, in addition to these analyses, with the analysis based on the inter-residue average distance statistics and our sequence tendency analysis, we could infer which part would play an important role in its structural formation.
Conclusions: The results suggest possible determinants of the different 3D structures for sequences with high sequence identity. The possibility of discriminating between the 3D structures based on the given sequences is also discussed.
Figures
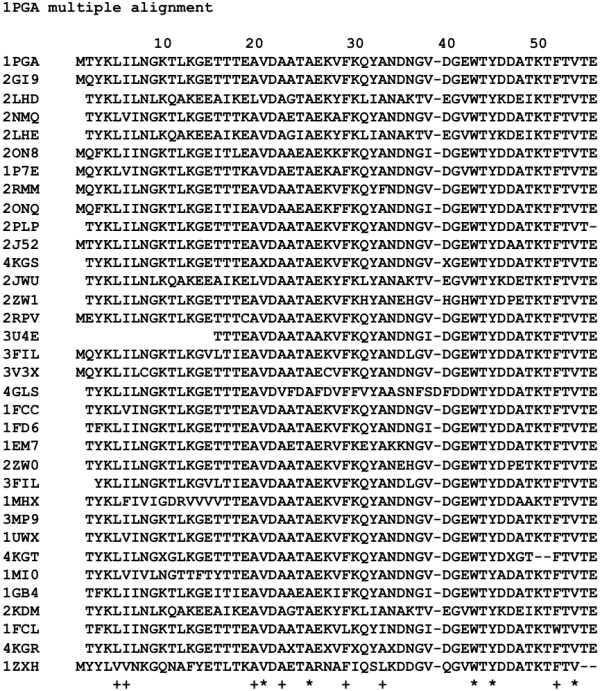
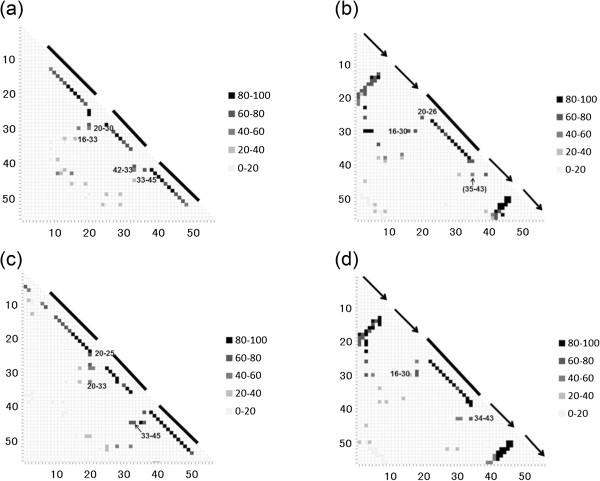
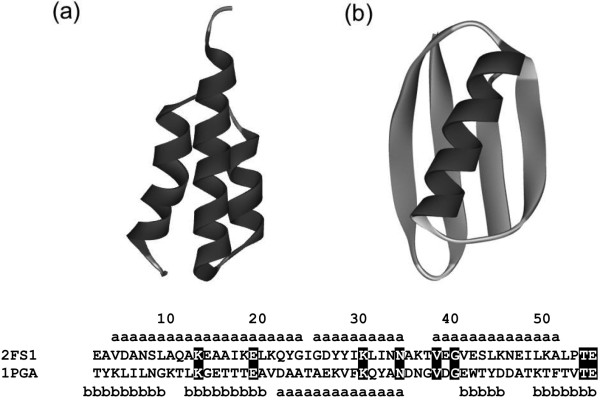
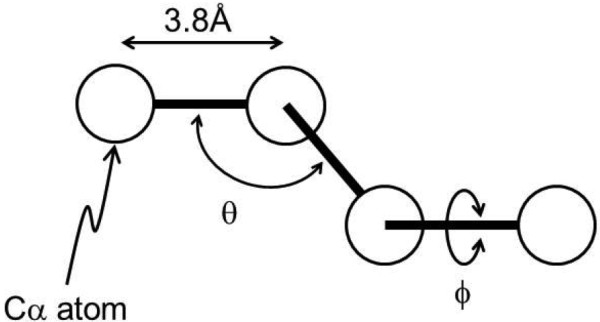
Similar articles
-
Analysis of 3D structural differences in the IgG-binding domains based on the interresidue average-distance statistics.Amino Acids. 2008 Oct;35(3):541-9. doi: 10.1007/s00726-008-0082-1. Epub 2008 Apr 23. Amino Acids. 2008. PMID: 18431532
-
NdPASA: a novel pairwise protein sequence alignment algorithm that incorporates neighbor-dependent amino acid propensities.Proteins. 2005 Feb 15;58(3):628-37. doi: 10.1002/prot.20359. Proteins. 2005. PMID: 15616964
-
An integrated approach to the analysis and modeling of protein sequences and structures. III. A comparative study of sequence conservation in protein structural families using multiple structural alignments.J Mol Biol. 2000 Aug 18;301(3):691-711. doi: 10.1006/jmbi.2000.3975. J Mol Biol. 2000. PMID: 10966778
-
[A turning point in the knowledge of the structure-function-activity relations of elastin].J Soc Biol. 2001;195(2):181-93. J Soc Biol. 2001. PMID: 11727705 Review. French.
-
Analyses of protein sequences using inter-residue average distance statistics to study folding processes and the significance of their partial sequences.Protein Pept Lett. 2011 Oct;18(10):979-90. doi: 10.2174/0929866511107010979. Protein Pept Lett. 2011. PMID: 21592083 Review.
Cited by
-
Conserved structural topologies in RNase A-like and trypsin-like serine proteases: a sequence-based folding analysis.BMC Mol Cell Biol. 2025 May 28;26(1):16. doi: 10.1186/s12860-025-00542-y. BMC Mol Cell Biol. 2025. PMID: 40437407 Free PMC article.
-
Analyses of the folding sites of irregular β-trefoil fold proteins through sequence-based techniques and Gō-model simulations.BMC Mol Cell Biol. 2020 Apr 15;21(1):28. doi: 10.1186/s12860-020-00271-4. BMC Mol Cell Biol. 2020. PMID: 32295515 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
