

A high-resolution network model for global gene regulation in Mycobacterium tuberculosis
- PMID: 25232098
- PMCID: PMC4191388
- DOI: 10.1093/nar/gku777
A high-resolution network model for global gene regulation in Mycobacterium tuberculosis
Abstract
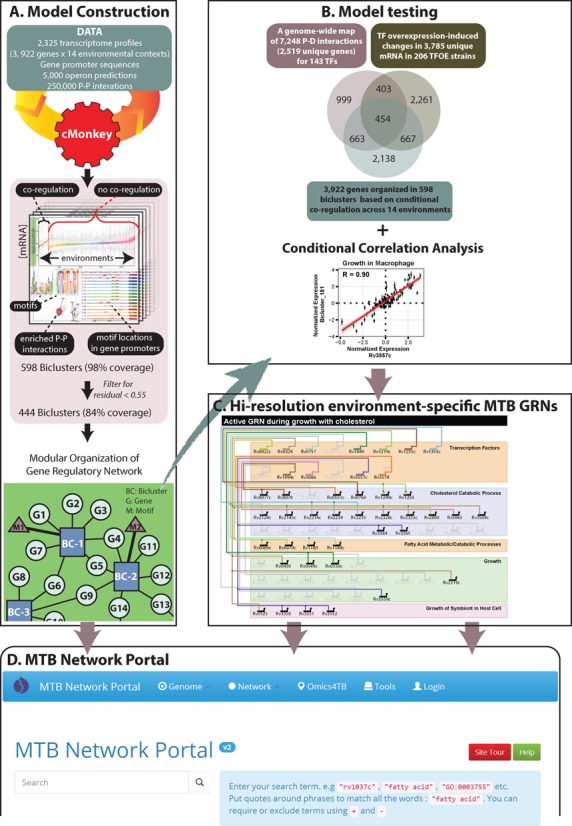
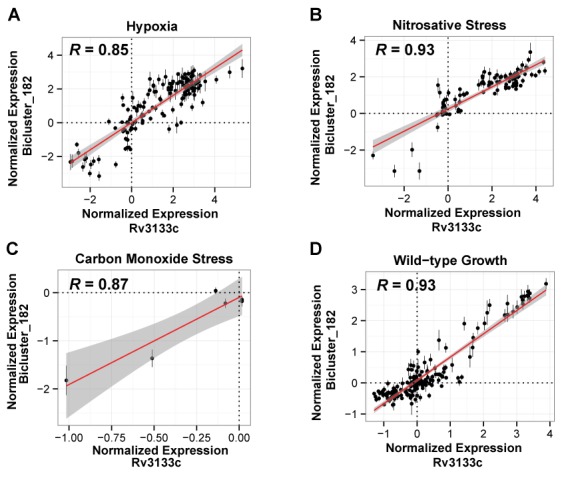
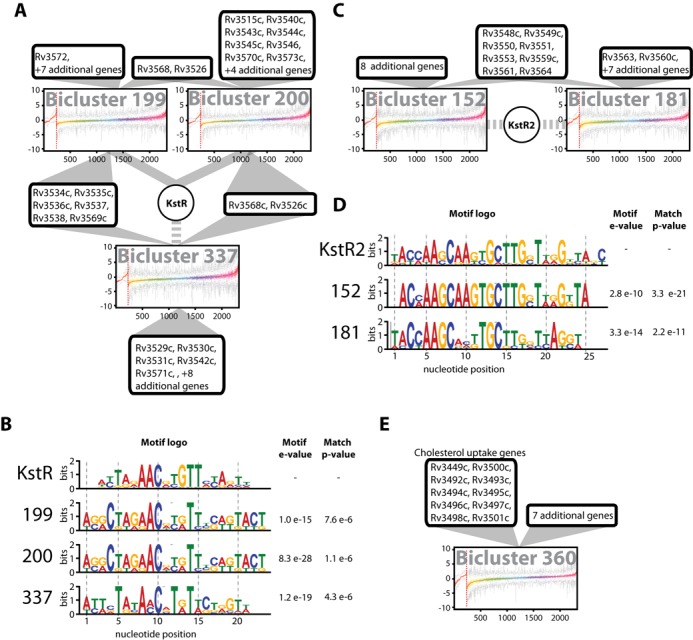
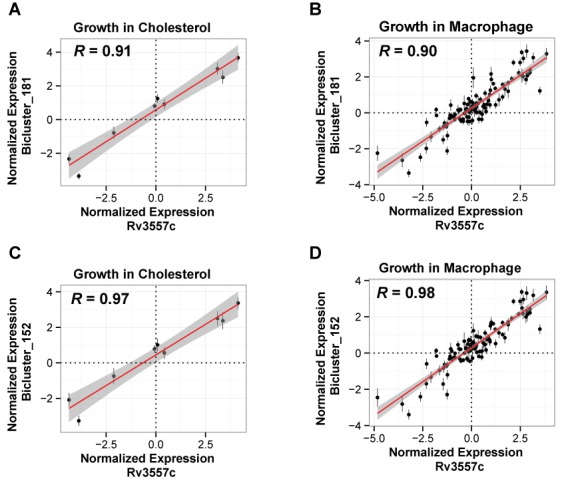
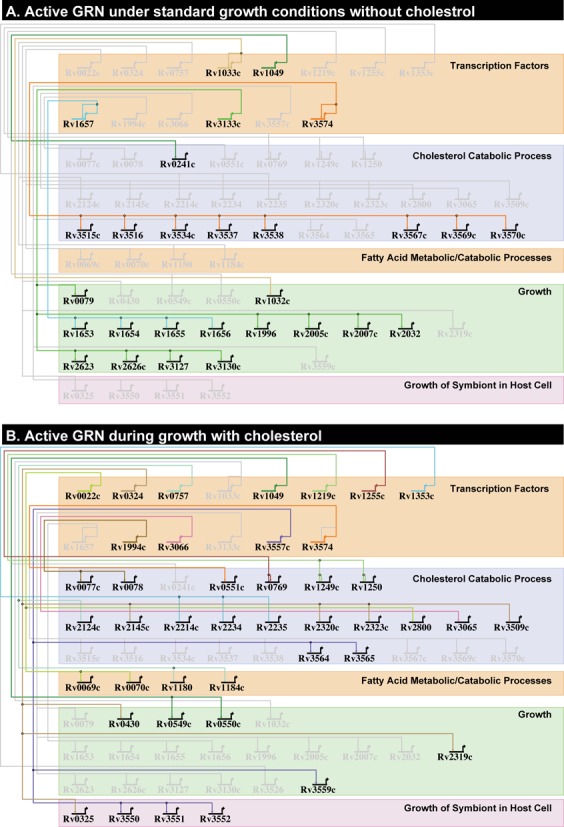
The resilience of Mycobacterium tuberculosis (MTB) is largely due to its ability to effectively counteract and even take advantage of the hostile environments of a host. In order to accelerate the discovery and characterization of these adaptive mechanisms, we have mined a compendium of 2325 publicly available transcriptome profiles of MTB to decipher a predictive, systems-scale gene regulatory network model. The resulting modular organization of 98% of all MTB genes within this regulatory network was rigorously tested using two independently generated datasets: a genome-wide map of 7248 DNA-binding locations for 143 transcription factors (TFs) and global transcriptional consequences of overexpressing 206 TFs. This analysis has discovered specific TFs that mediate conditional co-regulation of genes within 240 modules across 14 distinct environmental contexts. In addition to recapitulating previously characterized regulons, we discovered 454 novel mechanisms for gene regulation during stress, cholesterol utilization and dormancy. Significantly, 183 of these mechanisms act uniquely under conditions experienced during the infection cycle to regulate diverse functions including 23 genes that are essential to host-pathogen interactions. These and other insights underscore the power of a rational, model-driven approach to unearth novel MTB biology that operates under some but not all phases of infection.
© The Author(s) 2014. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
-
- Bonneau R., Facciotti M.T., Reiss D.J., Schmid A.K., Pan M., Kaur A., Thorsson V., Shannon P., Johnson M.H., Bare J.C., et al. A predictive model for transcriptional control of physiology in a free living cell. Cell. 2007;131:1354–1365. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
