

Cation-chloride cotransporters in neuronal development, plasticity and disease
- PMID: 25234263
- PMCID: PMC4294553
- DOI: 10.1038/nrn3819
Cation-chloride cotransporters in neuronal development, plasticity and disease
Abstract
Electrical activity in neurons requires a seamless functional coupling between plasmalemmal ion channels and ion transporters. Although ion channels have been studied intensively for several decades, research on ion transporters is in its infancy. In recent years, it has become evident that one family of ion transporters, cation-chloride cotransporters (CCCs), and in particular K(+)-Cl(-) cotransporter 2 (KCC2), have seminal roles in shaping GABAergic signalling and neuronal connectivity. Studying the functions of these transporters may lead to major paradigm shifts in our understanding of the mechanisms underlying brain development and plasticity in health and disease.
Conflict of interest statement
The authors declare no competing interests.
Figures
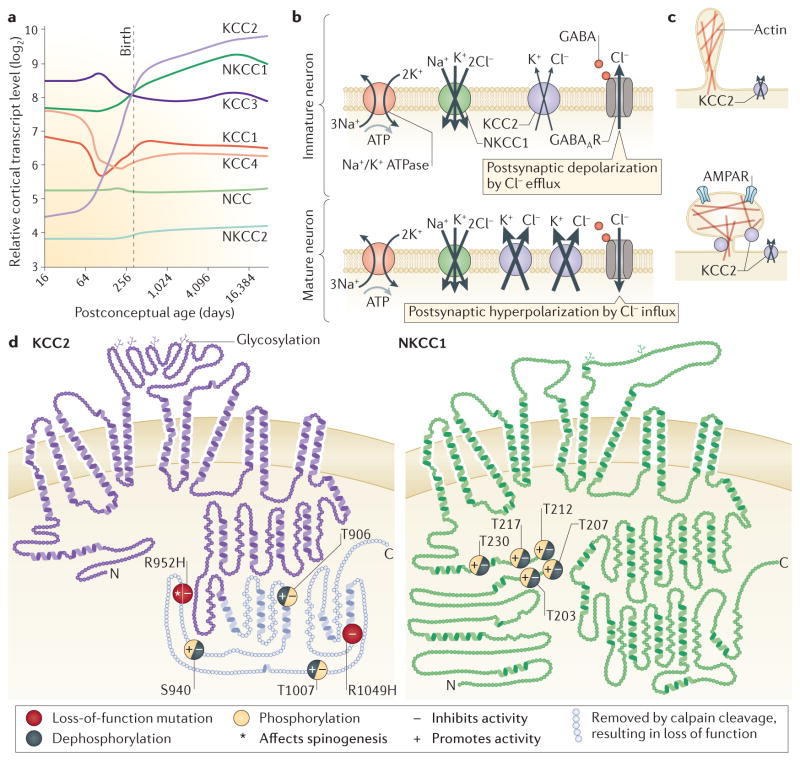
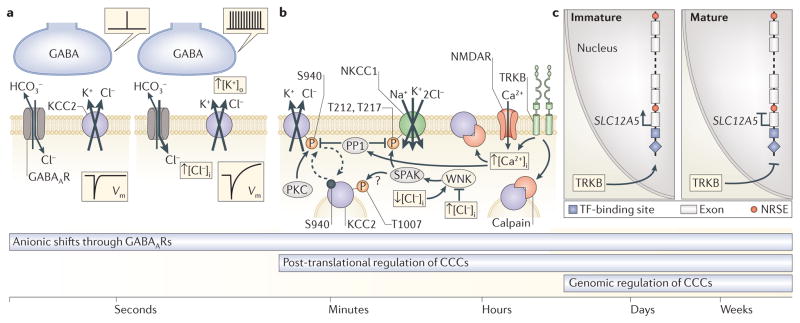

References
-
- Kaila K. Ionic basis of GABAA receptor channel function in the nervous system. Prog Neurobiol. 1994;42:489–537. - PubMed
-
- Rivera C, et al. The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature. 1999;397:251–255. This was the first study to demonstrate the causal role of KCC2 upregulation in the development of hyperpolarizing inhibition. - PubMed
-
- Stein V, Hermans-Borgmeyer I, Jentsch TJ, Hübner CA. Expression of the KCl cotransporter KCC2 parallels neuronal maturation and the emergence of low intracellular chloride. J Comp Neurol. 2004;468:57–64. - PubMed
-
- Zhu L, Lovinger D, Delpire E. Cortical neurons lacking KCC2 expression show impaired regulation of intracellular chloride. J Neurophysiol. 2005;93:1557–1568. - PubMed
-
- Seja P, et al. Raising cytosolic Cl− in cerebellar granule cells affects their excitability and vestibulo-ocular learning. EMBO J. 2012;31:1217–1230. This elegant study used cell-specific genetic targeting of KCC2 and KCC3 to clarify their contribution to Cl− regulation and spine morphology in the mature cerebellar cortex. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
