

Burkholderia pseudomallei sequencing identifies genomic clades with distinct recombination, accessory, and epigenetic profiles
- PMID: 25236617
- PMCID: PMC4317168
- DOI: 10.1101/gr.177543.114
Burkholderia pseudomallei sequencing identifies genomic clades with distinct recombination, accessory, and epigenetic profiles
Erratum in
-
Burkholderia pseudomallei sequencing identifies genomic clades with distinct recombination, accessory, and epigenetic profiles.Genome Res. 2015 Apr;25(4):608. Genome Res. 2015. PMID: 25834186 Free PMC article. No abstract available.
Abstract
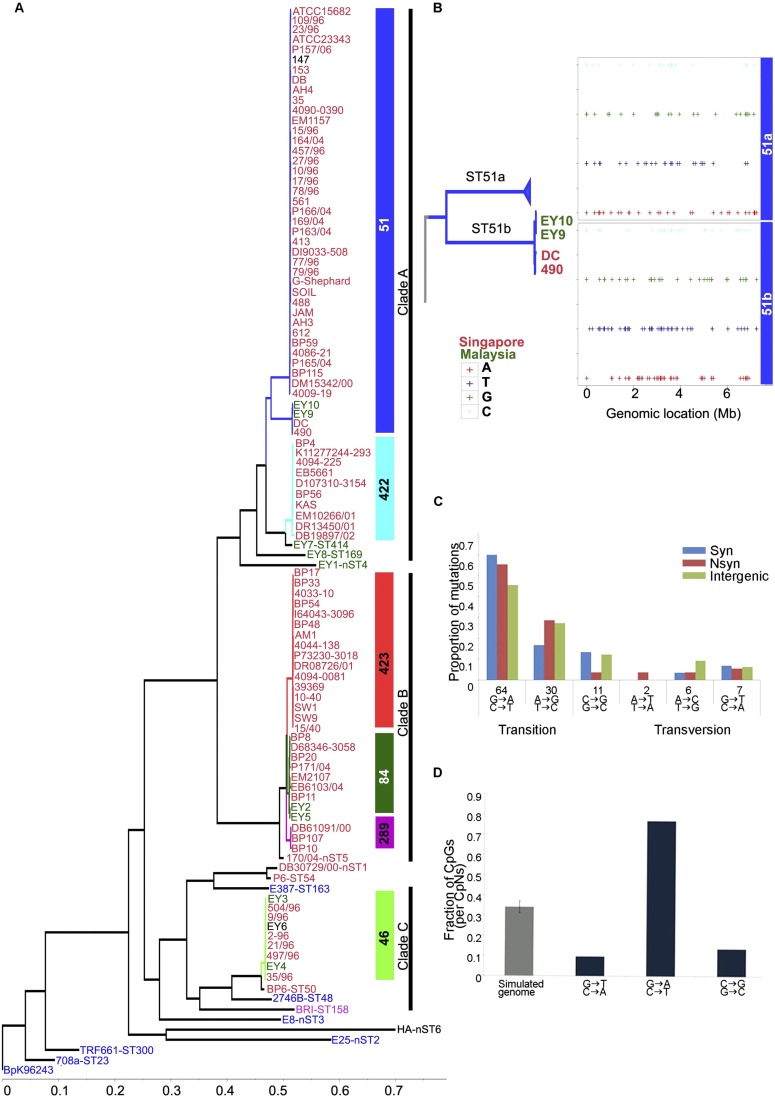
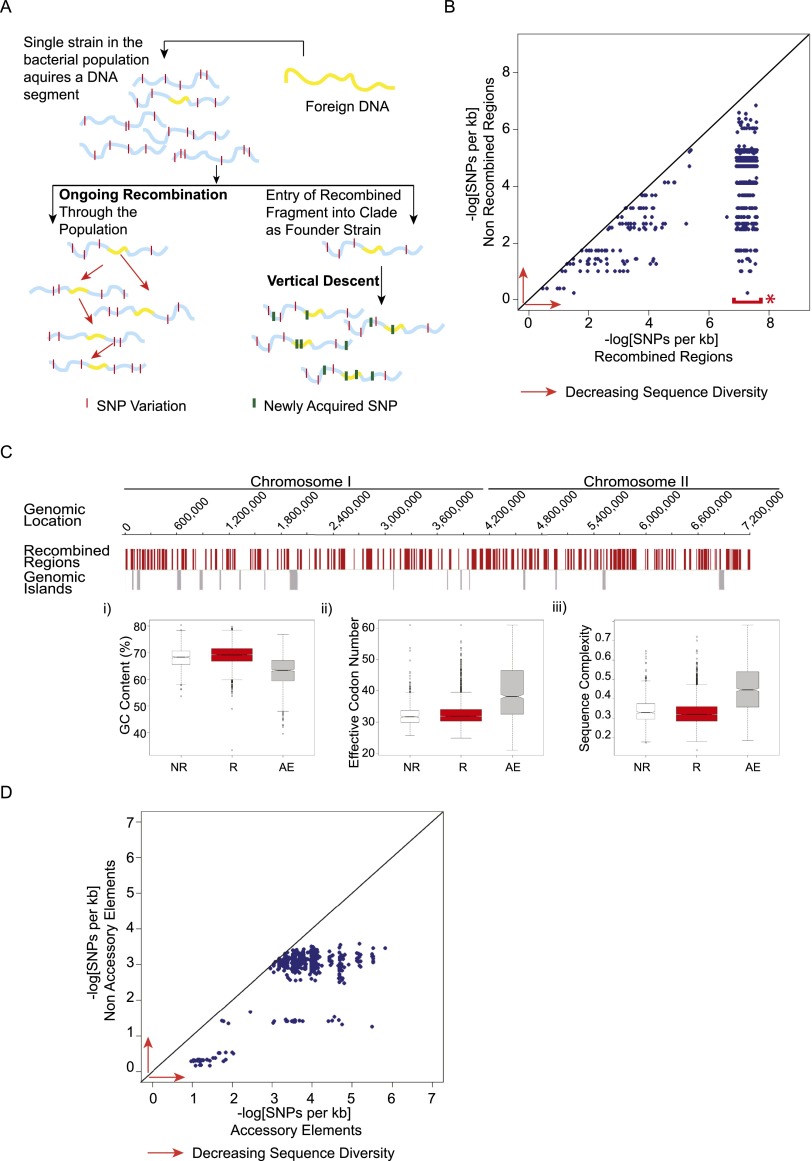
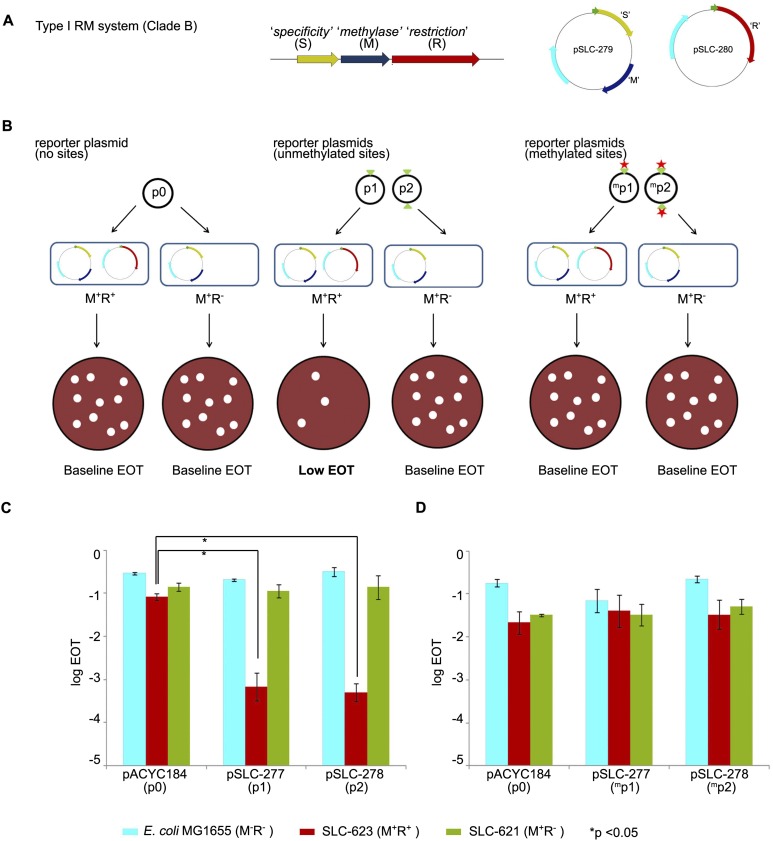
Burkholderia pseudomallei (Bp) is the causative agent of the infectious disease melioidosis. To investigate population diversity, recombination, and horizontal gene transfer in closely related Bp isolates, we performed whole-genome sequencing (WGS) on 106 clinical, animal, and environmental strains from a restricted Asian locale. Whole-genome phylogenies resolved multiple genomic clades of Bp, largely congruent with multilocus sequence typing (MLST). We discovered widespread recombination in the Bp core genome, involving hundreds of regions associated with multiple haplotypes. Highly recombinant regions exhibited functional enrichments that may contribute to virulence. We observed clade-specific patterns of recombination and accessory gene exchange, and provide evidence that this is likely due to ongoing recombination between clade members. Reciprocally, interclade exchanges were rarely observed, suggesting mechanisms restricting gene flow between clades. Interrogation of accessory elements revealed that each clade harbored a distinct complement of restriction-modification (RM) systems, predicted to cause clade-specific patterns of DNA methylation. Using methylome sequencing, we confirmed that representative strains from separate clades indeed exhibit distinct methylation profiles. Finally, using an E. coli system, we demonstrate that Bp RM systems can inhibit uptake of non-self DNA. Our data suggest that RM systems borne on mobile elements, besides preventing foreign DNA invasion, may also contribute to limiting exchanges of genetic material between individuals of the same species. Genomic clades may thus represent functional units of genetic isolation in Bp, modulating intraspecies genetic diversity.
© 2015 Nandi et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases