

Transforming growth factor β-mediated suppression of antitumor T cells requires FoxP1 transcription factor expression
- PMID: 25238097
- PMCID: PMC4174366
- DOI: 10.1016/j.immuni.2014.08.012
Transforming growth factor β-mediated suppression of antitumor T cells requires FoxP1 transcription factor expression
Abstract
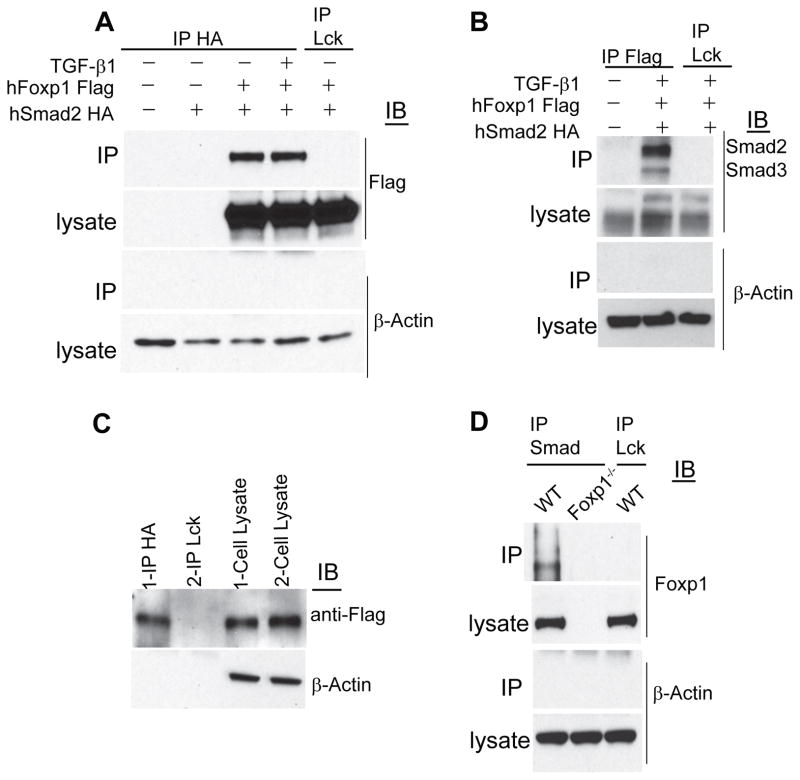
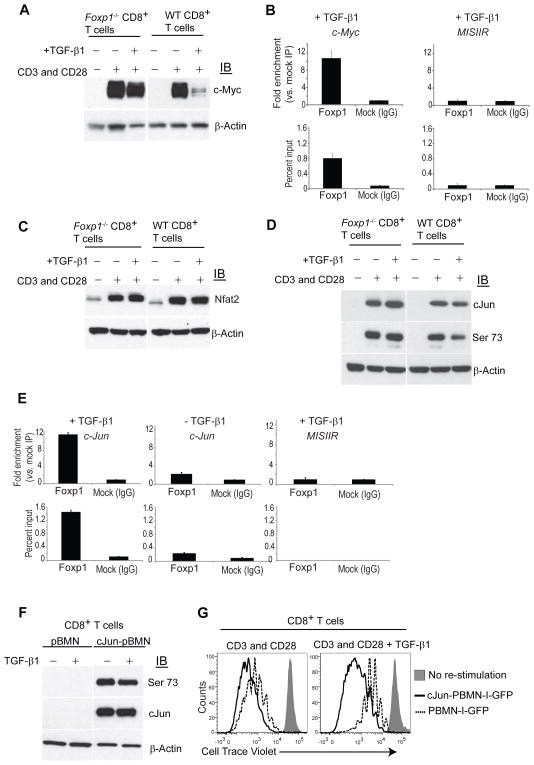
Tumor-reactive T cells become unresponsive in advanced tumors. Here we have characterized a common mechanism of T cell unresponsiveness in cancer driven by the upregulation of the transcription factor Forkhead box protein P1 (Foxp1), which prevents CD8⁺ T cells from proliferating and upregulating Granzyme-B and interferon-γ in response to tumor antigens. Accordingly, Foxp1-deficient lymphocytes induced rejection of incurable tumors and promoted protection against tumor rechallenge. Mechanistically, Foxp1 interacted with the transcription factors Smad2 and Smad3 in preactivated CD8⁺ T cells in response to microenvironmental transforming growth factor-β (TGF-β), and was essential for its suppressive activity. Therefore, Smad2 and Smad3-mediated c-Myc repression requires Foxp1 expression in T cells. Furthermore, Foxp1 directly mediated TGF-β-induced c-Jun transcriptional repression, which abrogated T cell activity. Our results unveil a fundamental mechanism of T cell unresponsiveness different from anergy or exhaustion, driven by TGF-β signaling on tumor-associated lymphocytes undergoing Foxp1-dependent transcriptional regulation.
Copyright © 2014 Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
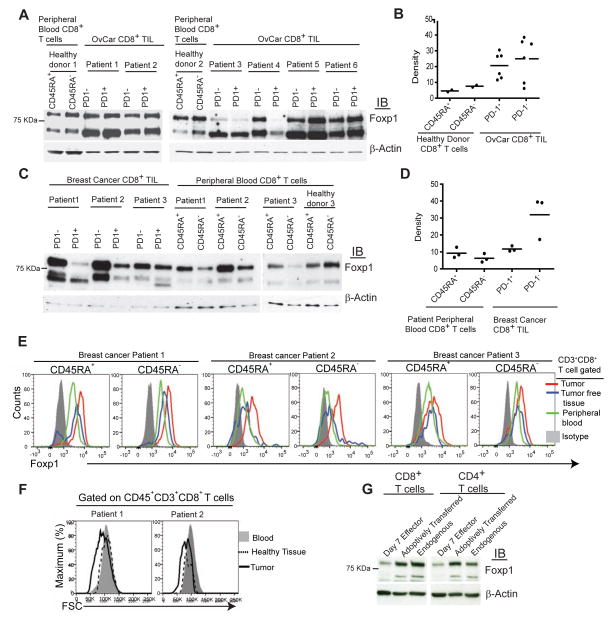
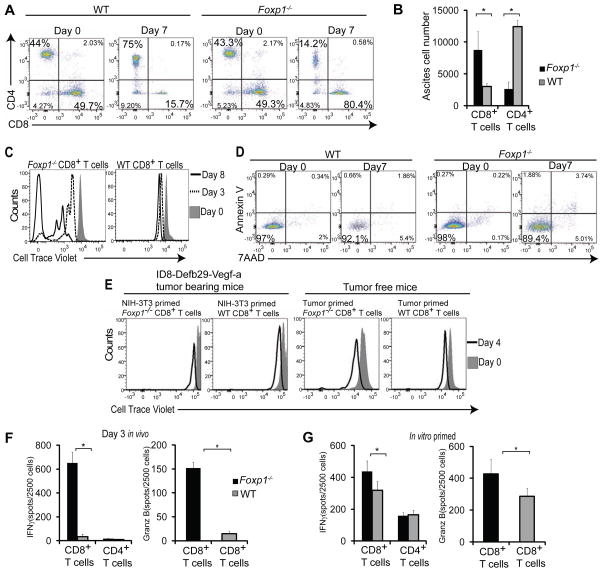
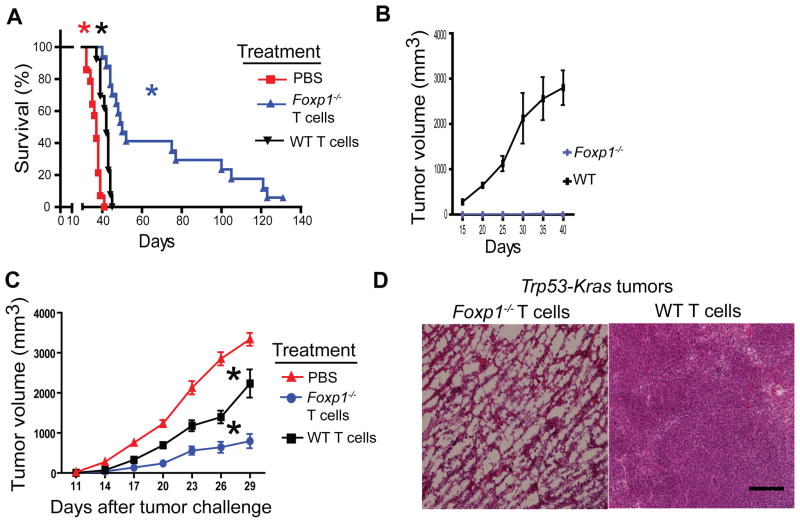
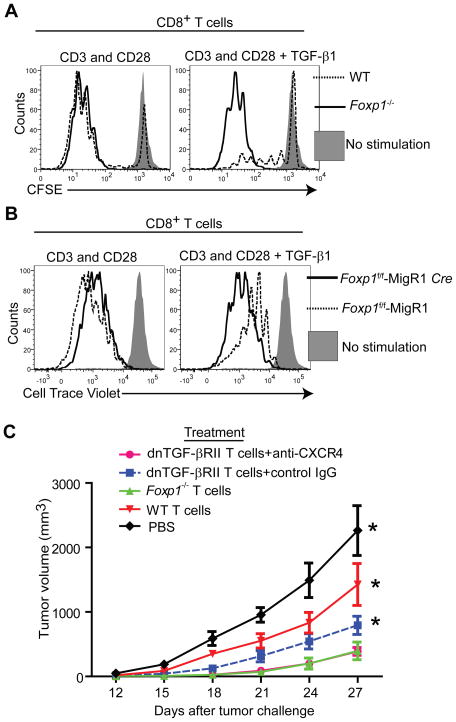

Comment in
-
Targeting foxp1 for reinstating anticancer immunosurveillance.Immunity. 2014 Sep 18;41(3):345-347. doi: 10.1016/j.immuni.2014.09.001. Immunity. 2014. PMID: 25238089
References
-
- Baitsch L, Fuertes-Marraco SA, Legat A, Meyer C, Speiser DE. The three main stumbling blocks for anticancer T cells. Trends Immunol 2012 - PubMed
-
- Chen L, Glover JN, Hogan PG, Rao A, Harrison SC. Structure of the DNA-binding domains from NFAT, Fos and Jun bound specifically to DNA. Nature. 1998;392:42–48. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
