

In vitro and in vivo interactions between the HDAC6 inhibitor ricolinostat (ACY1215) and the irreversible proteasome inhibitor carfilzomib in non-Hodgkin lymphoma cells
- PMID: 25239935
- PMCID: PMC4304772
- DOI: 10.1158/1535-7163.MCT-14-0220
In vitro and in vivo interactions between the HDAC6 inhibitor ricolinostat (ACY1215) and the irreversible proteasome inhibitor carfilzomib in non-Hodgkin lymphoma cells
Retraction in
-
Retraction: In Vitro and In Vivo Interactions between the HDAC6 Inhibitor Ricolinostat (ACY1215) and the Irreversible Proteasome Inhibitor Carfilzomib in Non-Hodgkin Lymphoma Cells.Mol Cancer Ther. 2019 Jun;18(6):1181. doi: 10.1158/1535-7163.MCT-19-0471. Mol Cancer Ther. 2019. PMID: 31160511 Free PMC article. No abstract available.
Abstract
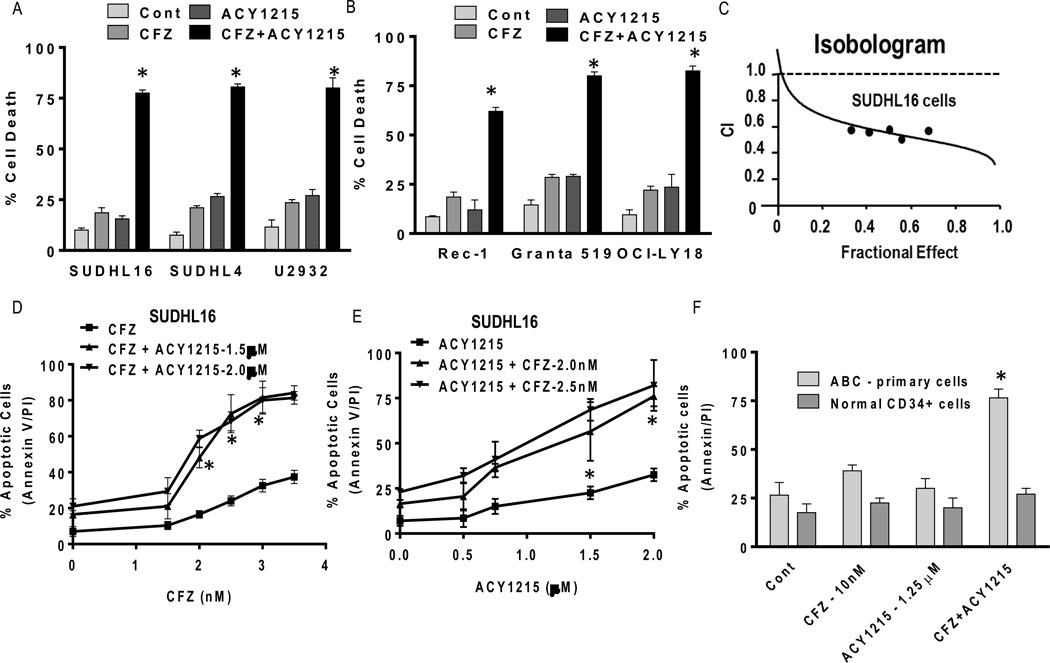
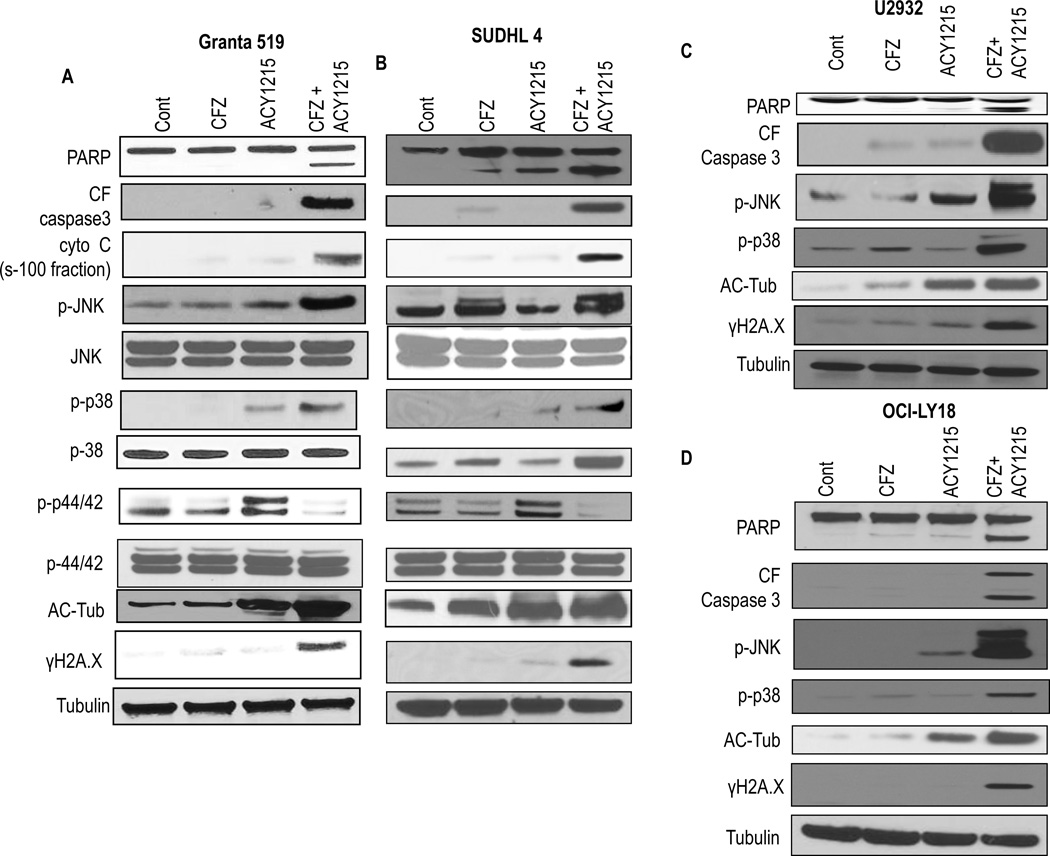
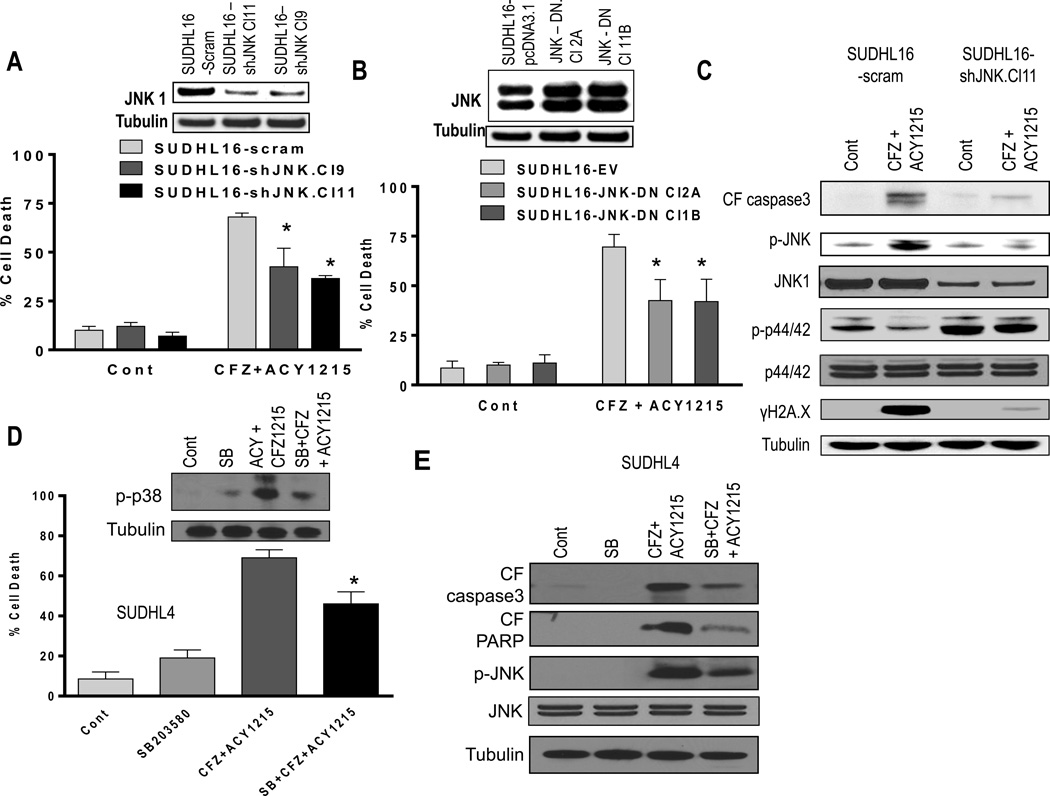
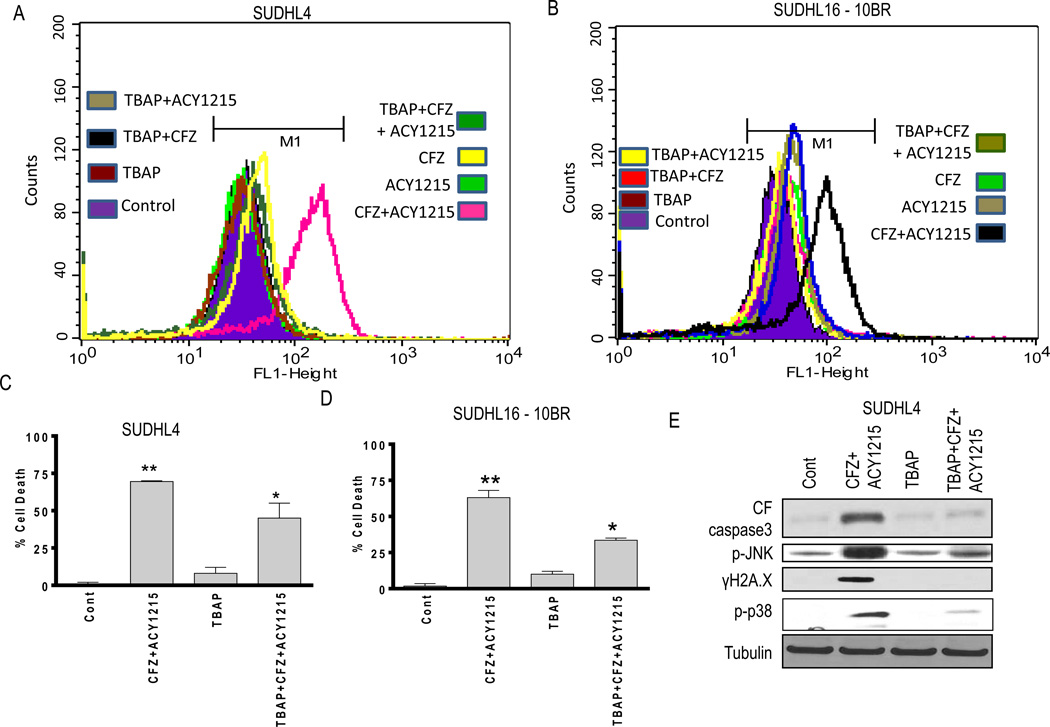
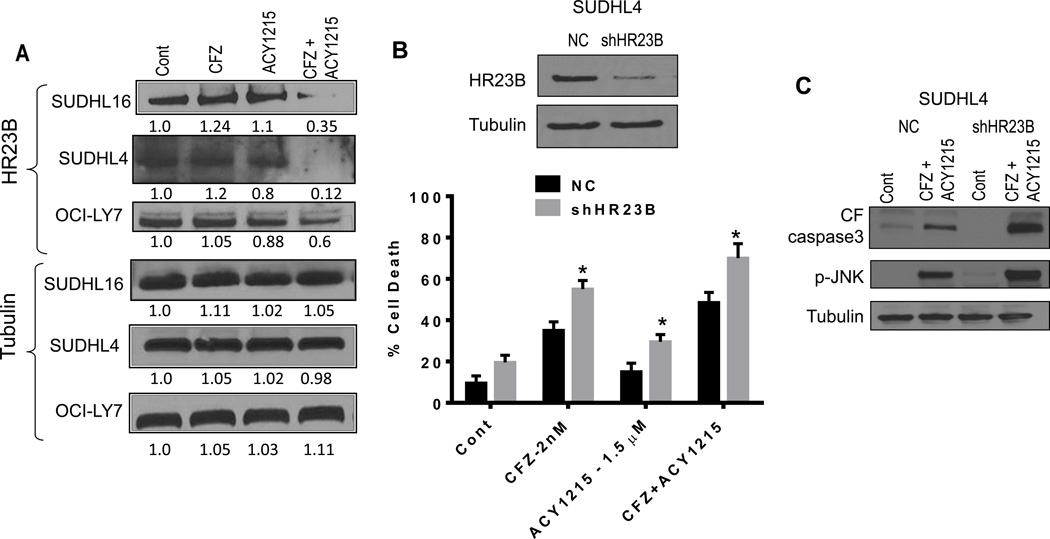
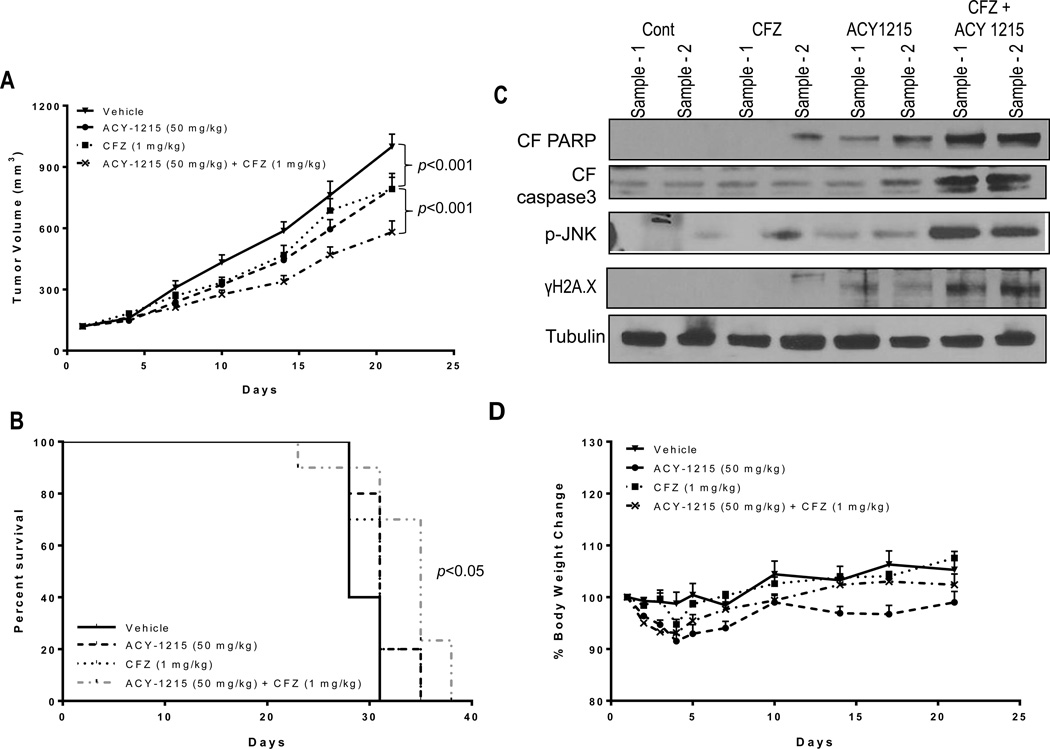
Interactions between the HDAC6 inhibitor ricolinostat (ACY1215) and the irreversible proteasome inhibitor carfilzomib were examined in non-Hodgkin lymphoma (NHL) models, including diffuse large B-cell lymphoma (DLBCL), mantle cell lymphoma (MCL), and double-hit lymphoma cells. Marked in vitro synergism was observed in multiple cell types associated with activation of cellular stress pathways (e.g., JNK1/2, ERK1/2, and p38) accompanied by increases in DNA damage (γH2A.X), G2-M arrest, and the pronounced induction of mitochondrial injury and apoptosis. Combination treatment with carfilzomib and ricolinostat increased reactive oxygen species (ROS), whereas the antioxidant TBAP attenuated DNA damage, JNK activation, and cell death. Similar interactions occurred in bortezomib-resistant and double-hit DLBCL, MCL, and primary DLBCL cells, but not in normal CD34(+) cells. However, ricolinostat did not potentiate inhibition of chymotryptic activity by carfilzomib. shRNA knockdown of JNK1 (but not MEK1/2), or pharmacologic inhibition of p38, significantly reduced carfilzomib-ricolinostat lethality, indicating a functional contribution of these stress pathways to apoptosis. Combined exposure to carfilzomib and ricolinostat also markedly downregulated the cargo-loading protein HR23B. Moreover, HR23B knockdown significantly increased carfilzomib- and ricolinostat-mediated lethality, suggesting a role for this event in cell death. Finally, combined in vivo treatment with carfilzomib and ricolinostat was well tolerated and significantly suppressed tumor growth and increased survival in an MCL xenograft model. Collectively, these findings indicate that carfilzomib and ricolinostat interact synergistically in NHL cells through multiple stress-related mechanisms, and suggest that this strategy warrants further consideration in NHL.
©2014 American Association for Cancer Research.
Conflict of interest statement
Potential conflict of interest: SNQ and SSJ are employees of Acetylon Pharmaceuticals, Inc. All other authors have no conflicts of interest to report
Figures
Comment in
-
Findings of Research Misconduct.NIH Guide Grants Contracts (Bethesda). 2015 Dec 18:NOT-OD-16-040. NIH Guide Grants Contracts (Bethesda). 2015. PMID: 26693581 Free PMC article. No abstract available.
-
Findings of Research Misconduct.Fed Regist. 2015 Dec 10;80(237):76703-76704. Fed Regist. 2015. PMID: 27737268 Free PMC article. No abstract available.
References
-
- Friedberg JW. Double-hit diffuse large B-cell lymphoma. J Clin Oncol. 2012;30:3439–3443. - PubMed
-
- Schmidt C, Dreyling M. Therapy of mantle cell lymphoma: current standards and future strategies. Hematol Oncol Clin North Am. 2008;22:953–963. - PubMed
-
- Perez-Galan P, Roue G, Villamor N, Montserrat E, Campo E, Colomer D. The proteasome inhibitor bortezomib induces apoptosis in mantle-cell lymphoma through generation of ROS and Noxa activation independent of p53 status. Blood. 2006;107:257–264. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
