

Understanding the enormous diversity of bacteriophages: the tailed phages that infect the bacterial family Enterobacteriaceae
- PMID: 25240328
- PMCID: PMC4301999
- DOI: 10.1016/j.virol.2014.08.024
Understanding the enormous diversity of bacteriophages: the tailed phages that infect the bacterial family Enterobacteriaceae
Abstract
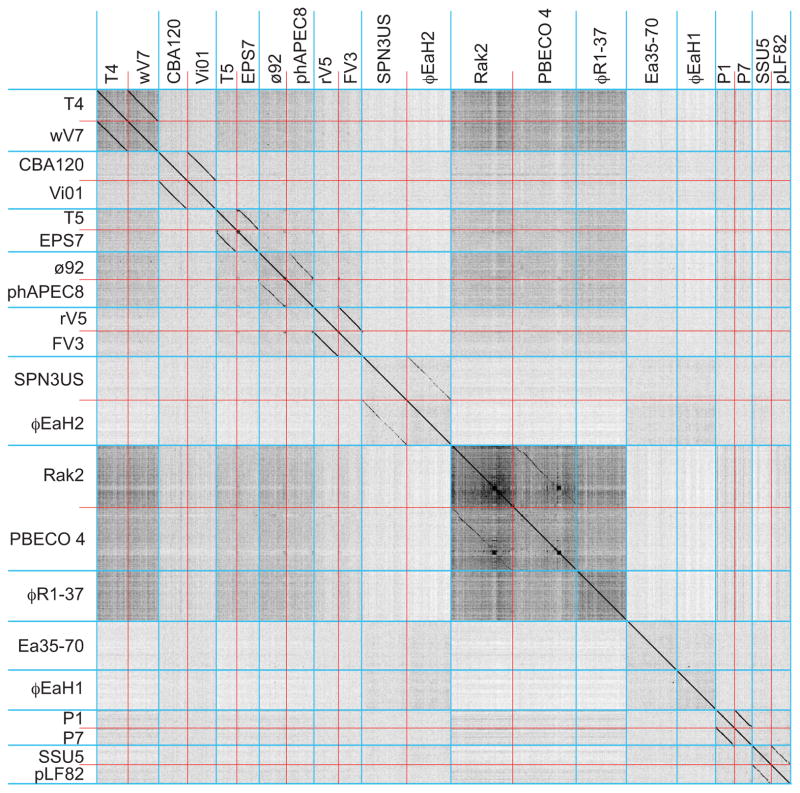
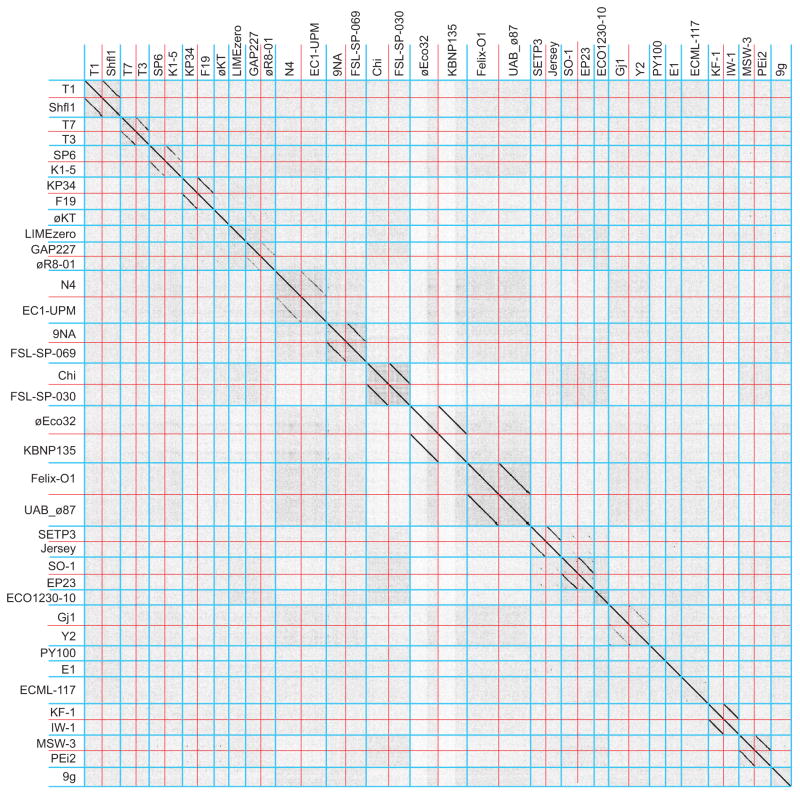
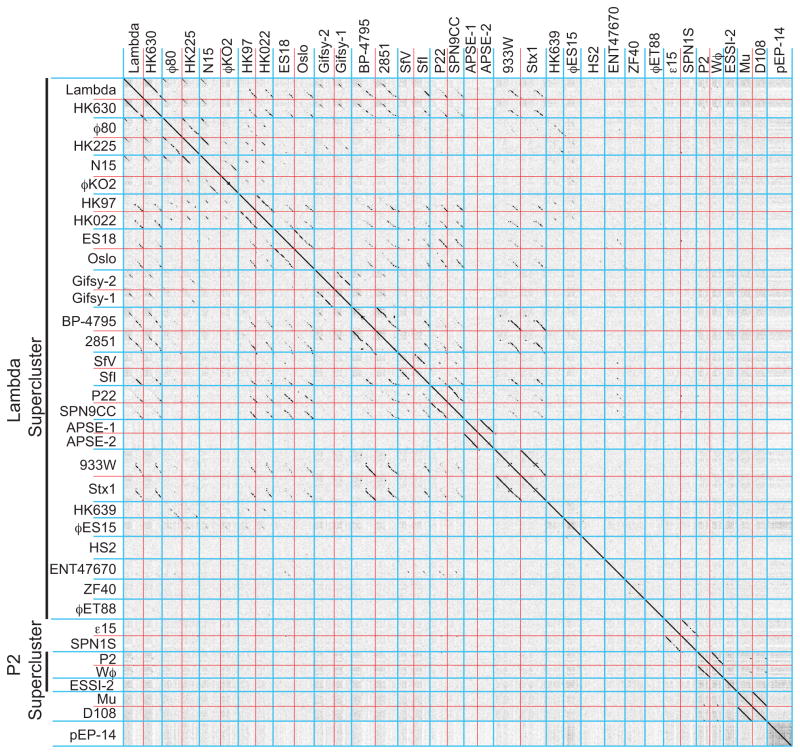
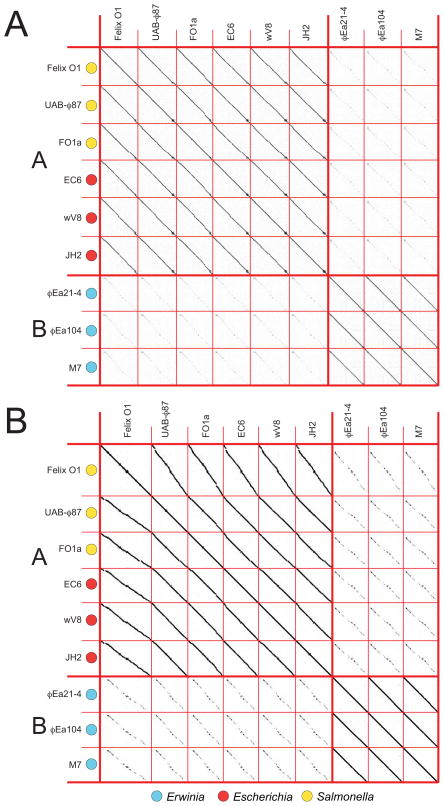
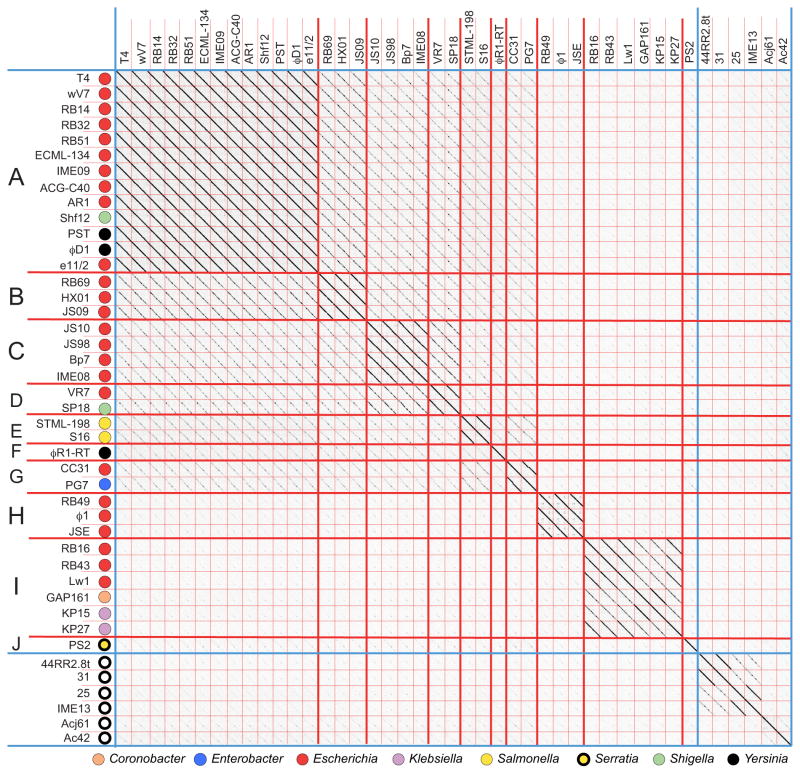
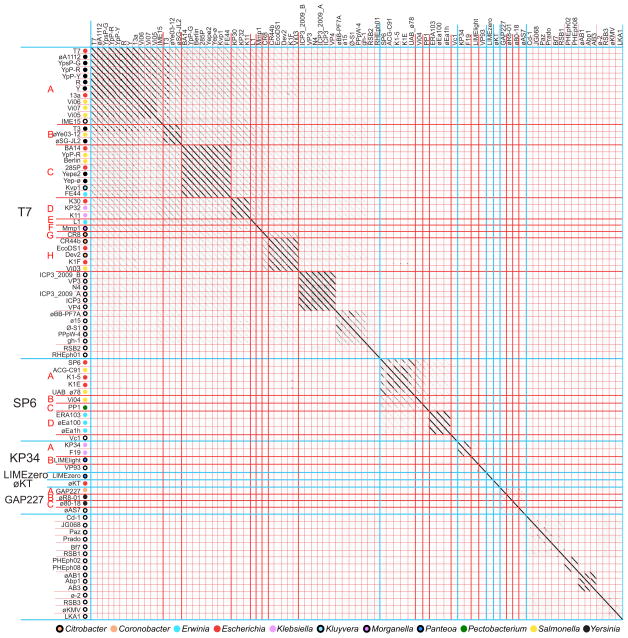
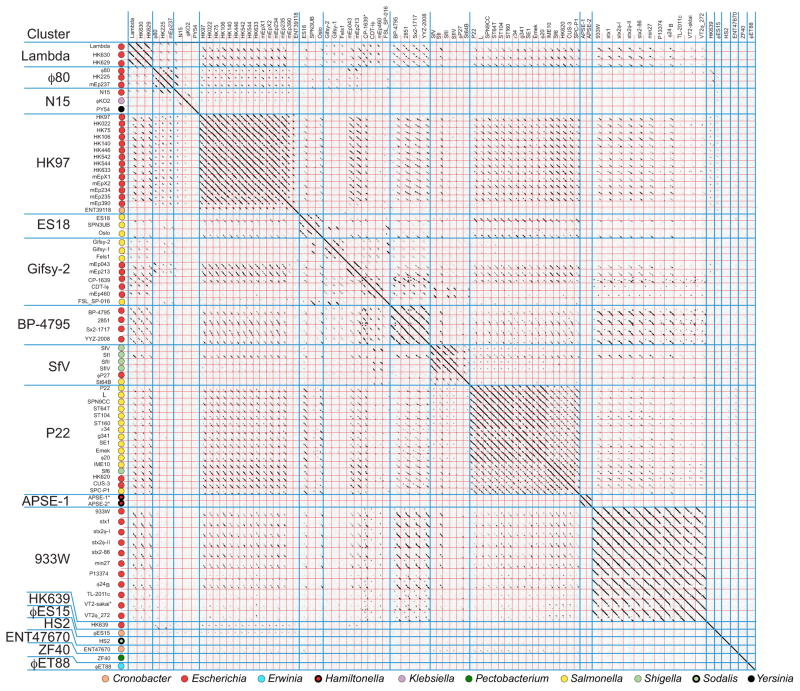
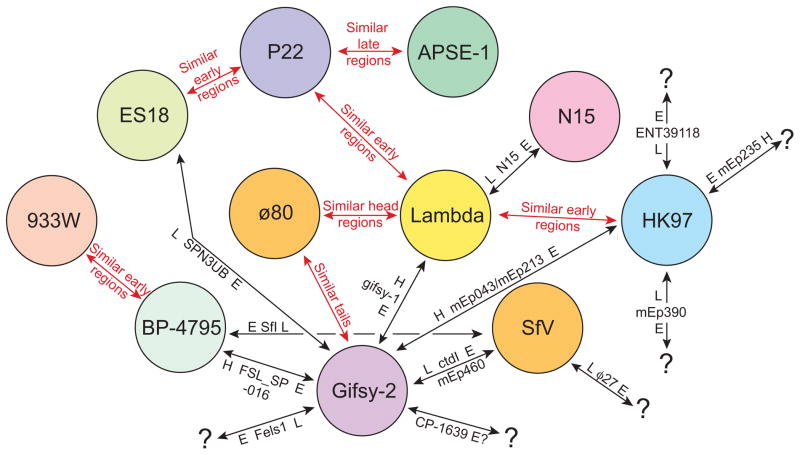
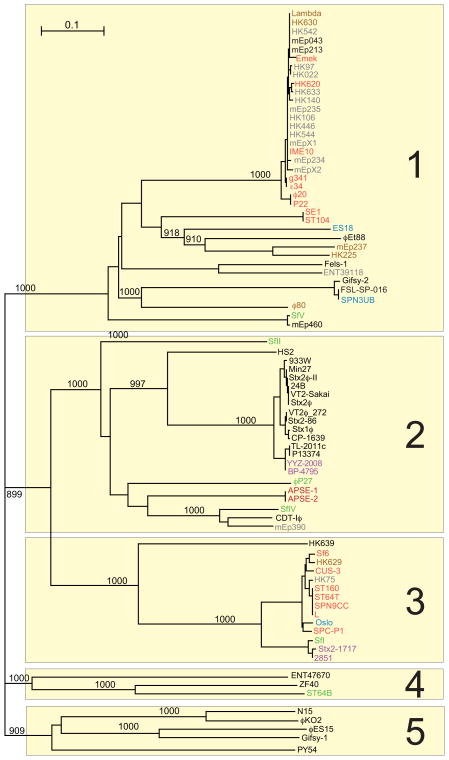
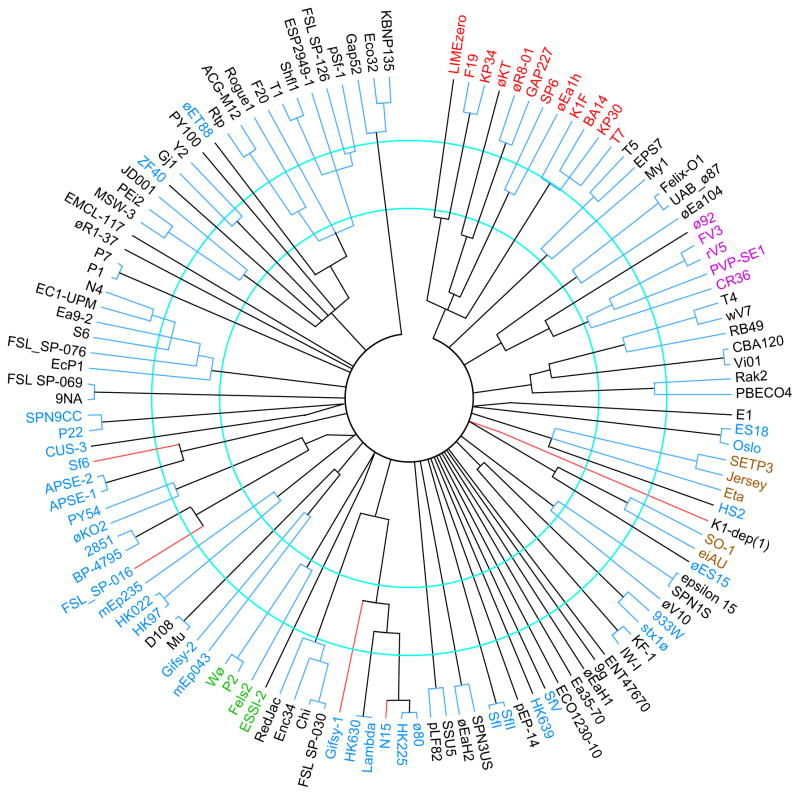
Bacteriophages are the predominant biological entity on the planet. The recent explosion of sequence information has made estimates of their diversity possible. We describe the genomic comparison of 337 fully sequenced tailed phages isolated on 18 genera and 31 species of bacteria in the Enterobacteriaceae. These phages were largely unambiguously grouped into 56 diverse clusters (32 lytic and 24 temperate) that have syntenic similarity over >50% of the genomes within each cluster, but substantially less sequence similarity between clusters. Most clusters naturally break into sets of more closely related subclusters, 78% of which are correlated with their host genera. The largest groups of related phages are superclusters united by genome synteny to lambda (81 phages) and T7 (51 phages). This study forms a robust framework for understanding diversity and evolutionary relationships of existing tailed phages, for relating newly discovered phages and for determining host/phage relationships.
Figures
References
-
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10. - PubMed
-
- Anderson TF. The activity of a bacteriostatic substance in the reaction between bacterial virus and host. Science. 1945;101:565–6. - PubMed
-
- Andres D, Roske Y, Doering C, Heinemann U, Seckler R, Barbirz S. Tail morphology controls DNA release in two Salmonella phages with one lipopolysaccharide receptor recognition system. Mol Microbiol. 2012;83:1244–53. - PubMed
-
- Baker J, Limberger R, Schneider SJ, Campbell A. Recombination and modular exchange in the genesis of new lambdoid phages. New Biol. 1991;3:297–308. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
