

The adaptive biasing force method: everything you always wanted to know but were afraid to ask
- PMID: 25247823
- PMCID: PMC4306294
- DOI: 10.1021/jp506633n
The adaptive biasing force method: everything you always wanted to know but were afraid to ask
Abstract
In the host of numerical schemes devised to calculate free energy differences by way of geometric transformations, the adaptive biasing force algorithm has emerged as a promising route to map complex free-energy landscapes. It relies upon the simple concept that as a simulation progresses, a continuously updated biasing force is added to the equations of motion, such that in the long-time limit it yields a Hamiltonian devoid of an average force acting along the transition coordinate of interest. This means that sampling proceeds uniformly on a flat free-energy surface, thus providing reliable free-energy estimates. Much of the appeal of the algorithm to the practitioner is in its physically intuitive underlying ideas and the absence of any requirements for prior knowledge about free-energy landscapes. Since its inception in 2001, the adaptive biasing force scheme has been the subject of considerable attention, from in-depth mathematical analysis of convergence properties to novel developments and extensions. The method has also been successfully applied to many challenging problems in chemistry and biology. In this contribution, the method is presented in a comprehensive, self-contained fashion, discussing with a critical eye its properties, applicability, and inherent limitations, as well as introducing novel extensions. Through free-energy calculations of prototypical molecular systems, many methodological aspects are examined, from stratification strategies to overcoming the so-called hidden barriers in orthogonal space, relevant not only to the adaptive biasing force algorithm but also to other importance-sampling schemes. On the basis of the discussions in this paper, a number of good practices for improving the efficiency and reliability of the computed free-energy differences are proposed.
Figures
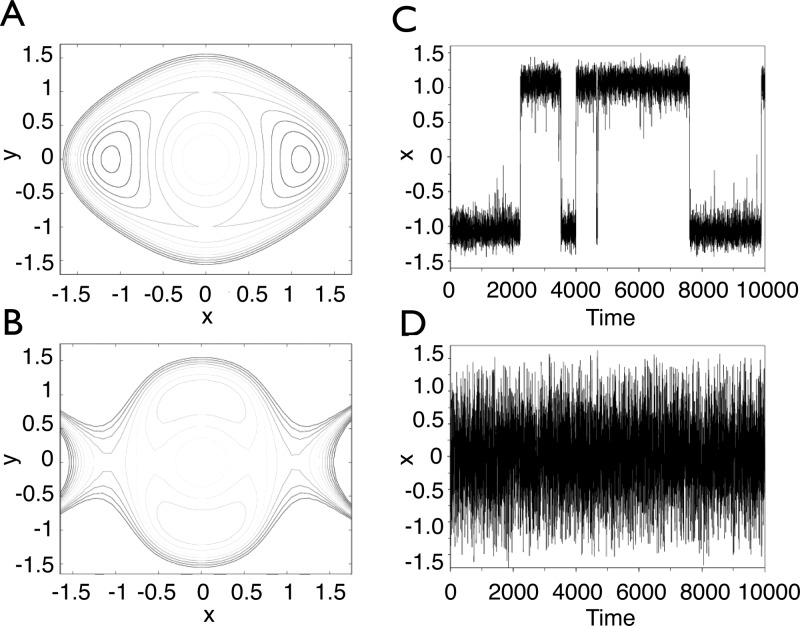
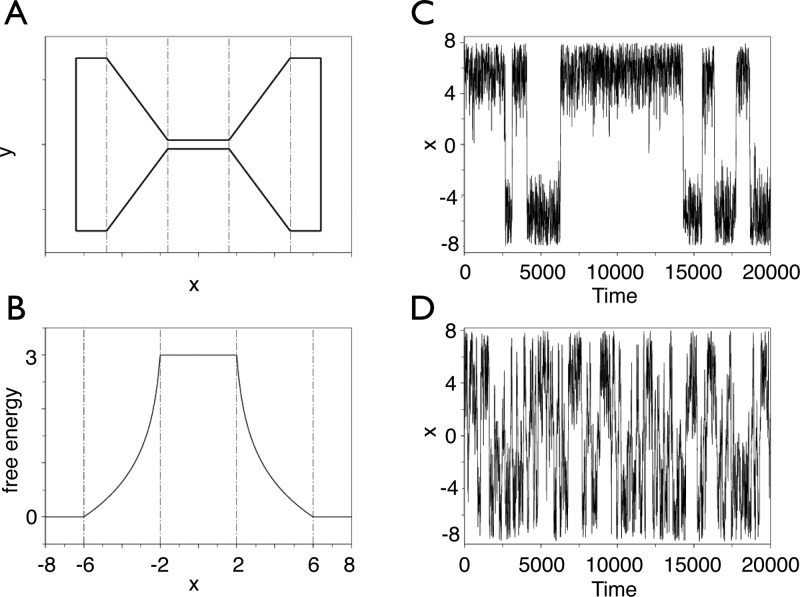
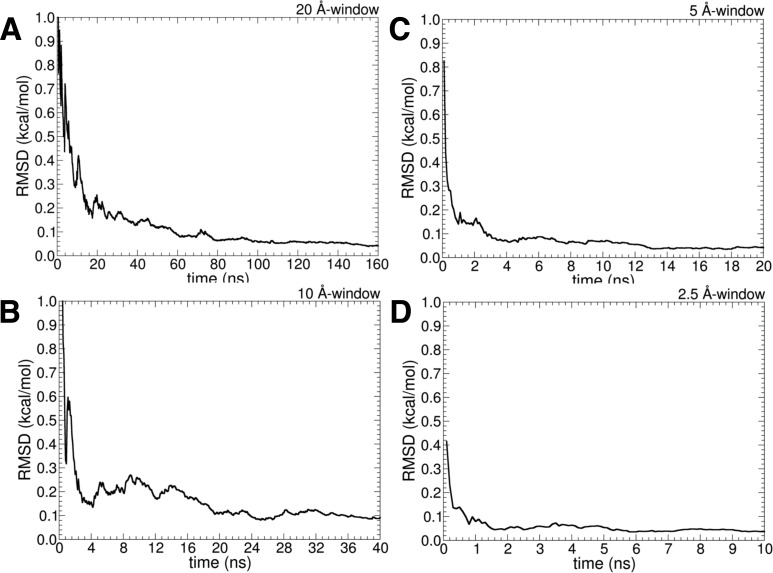
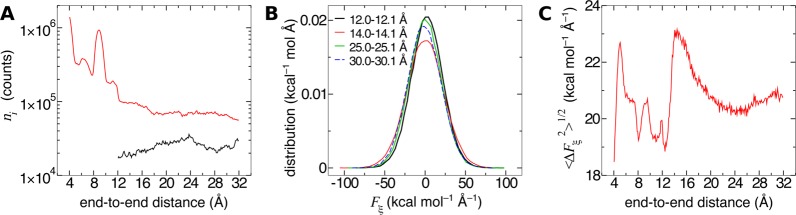
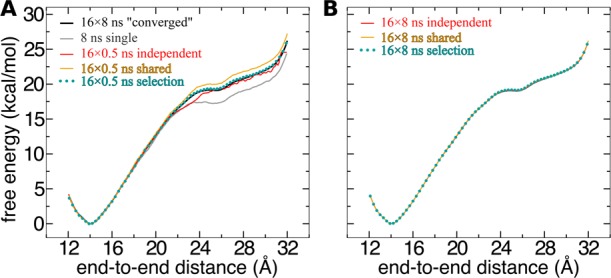
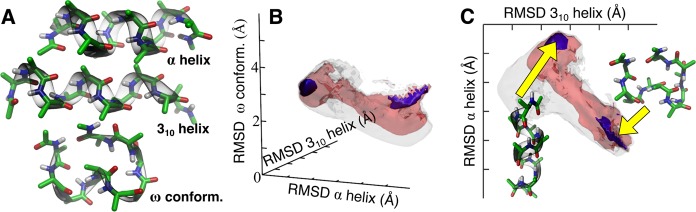
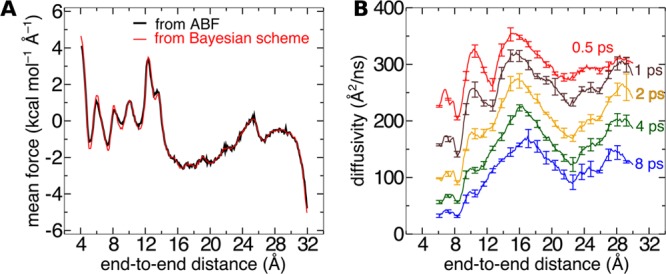
References
-
- Kirkwood J. G. Statistical Mechanics of Fluid Mixtures. J. Chem. Phys. 1935, 3, 300–313.
-
- Landau L. D.Statistical Physics; The Clarendon Press: Oxford, U.K., 1938.
-
- Zwanzig R. W. High-Temperature Equation of State by a Perturbation Method. I. Nonpolar Gases. J. Chem. Phys. 1954, 22, 1420–1426.
-
- McDonald I. R.; Singer K. Machine Calculation of Thermodynamic Properties of a Simple Fluid at Supercritical Temperatures. J. Chem. Phys. 1967, 47, 4766–4772.
-
- McDonald I. R.; Singer K. Calculation of Thermodynamic Properties of Liquid Argon from Lennard–jones Parameters by a Monte Carlo Method. Discuss. Faraday Soc. 1967, 43, 40–49.
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
