

Muscarinic cholinergic receptors modulate inhibitory synaptic rhythms in hippocampus and neocortex
- PMID: 25249974
- PMCID: PMC4155787
- DOI: 10.3389/fnsyn.2014.00018
Muscarinic cholinergic receptors modulate inhibitory synaptic rhythms in hippocampus and neocortex
Abstract
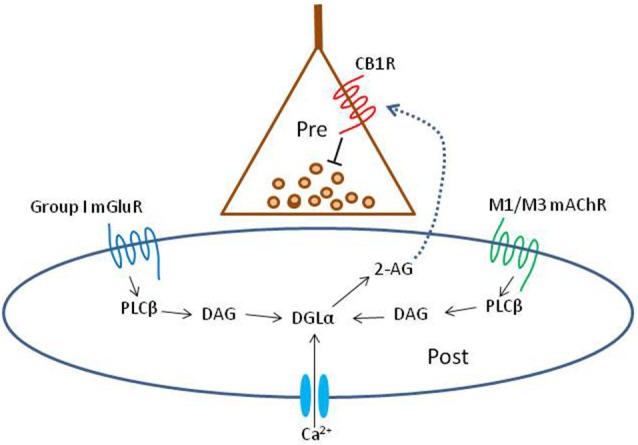
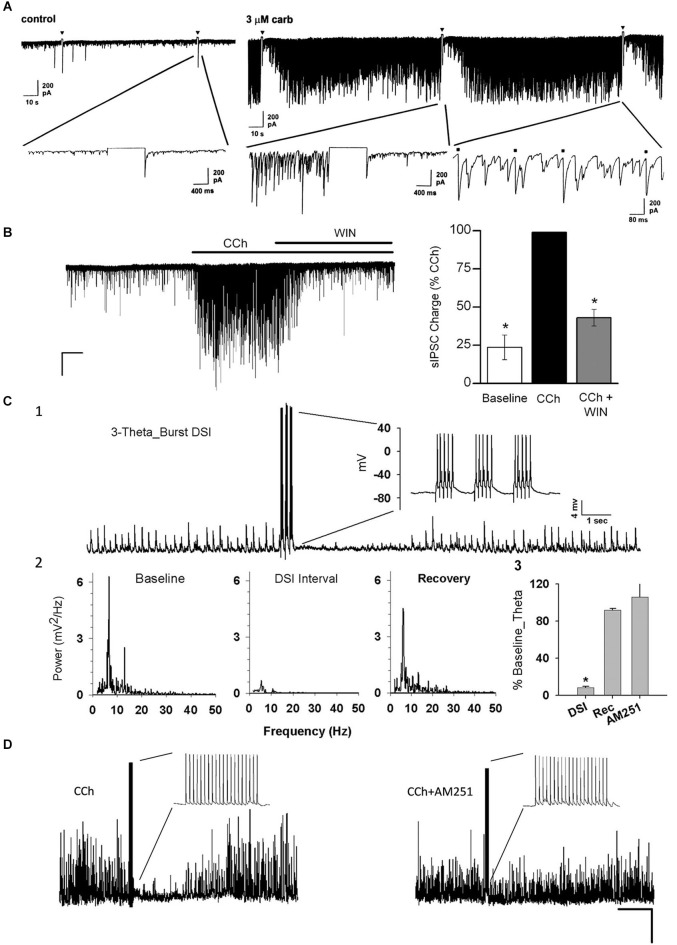
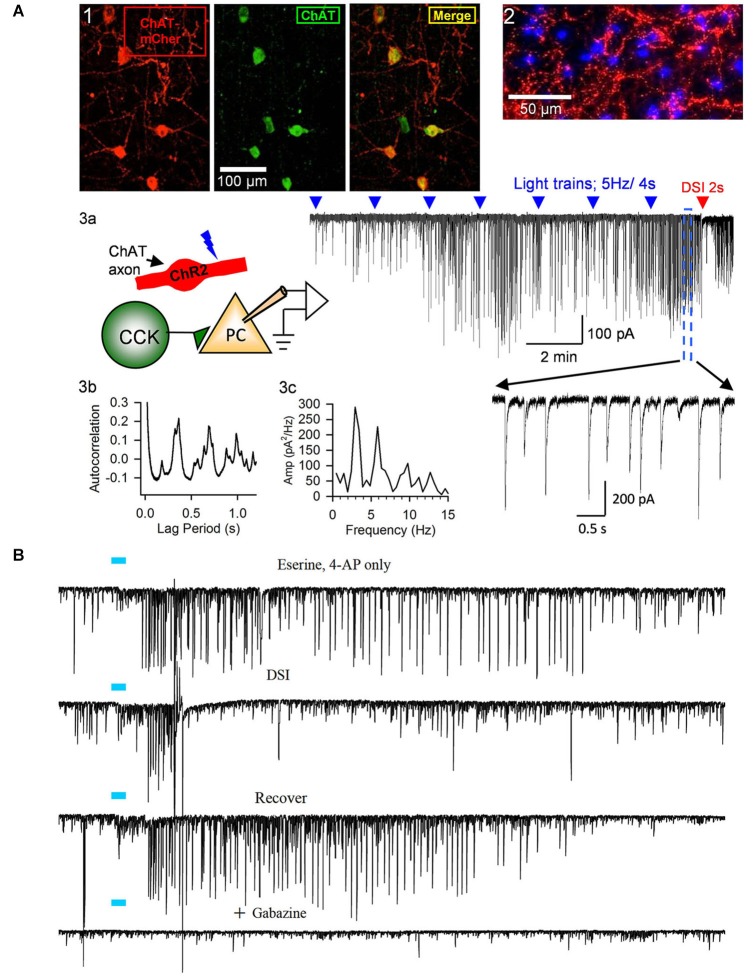
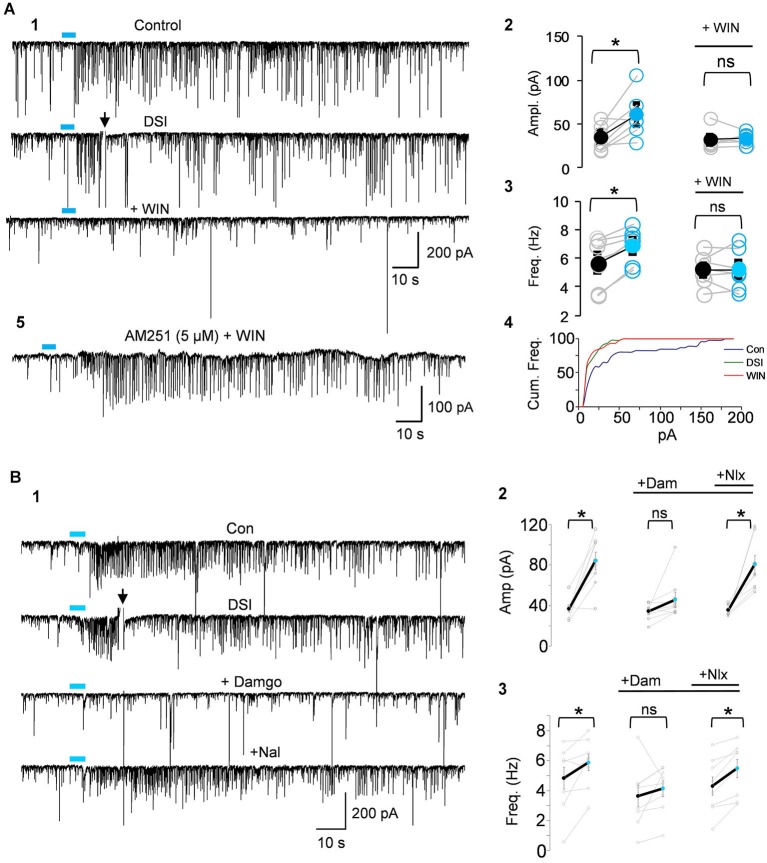
Activation of muscarinic acetylcholine (ACh) receptors (mAChRs) powerfully affects many neuronal properties as well as numerous cognitive behaviors. Small neuronal circuits constitute an intermediate level of organization between neurons and behaviors, and mAChRs affect interactions among cells that compose these circuits. Circuit activity is often assessed by extracellular recordings of the local field potentials (LFPs), which are analogous to in vivo EEGs, generated by coordinated neuronal interactions. Coherent forms of physiologically relevant circuit activity manifest themselves as rhythmic oscillations in the LFPs. Frequencies of rhythmic oscillations that are most closely associated with animal behavior are in the range of 4-80 Hz, which is subdivided into theta (4-14 Hz), beta (15-29 Hz) and gamma (30-80 Hz) bands. Activation of mAChRs triggers rhythmic oscillations in these bands in the hippocampus and neocortex. Inhibitory responses mediated by GABAergic interneurons constitute a prominent feature of these oscillations, and indeed, appear to be their major underlying factor in many cases. An important issue is which interneurons are involved in rhythm generation. Besides affecting cellular and network properties directly, mAChRs can cause the mobilization of endogenous cannabinoids (endocannabinoids, eCBs) that, by acting on the principal cannabinoid receptor of the brain, CB1R, regulate the release of certain neurotransmitters, including GABA. CB1Rs are heavily expressed on only a subset of interneurons and, at lower density, on glutamatergic neurons. Exogenous cannabinoids typically disrupt oscillations in the theta (θ) and gamma (γ) ranges, which probably contributes to the behavioral effects of these drugs. It is important to understand how neuronal circuit activity is affected by mAChR-driven eCBs, as this information will provide deeper insight into the actions of ACh itself, as well as into the effects of eCBs and exogenous cannabinoids in animal behavior. After covering some basic aspects of the mAChR system, this review will focus on recent findings concerning the mechanisms and circuitry that generate θ and γ rhythms in hippocampus and neocortex. The ability of optogenetic methods to probe the many roles of ACh in rhythm generation is highlighted.
Keywords: GABA; cholecystokinin; endocannabinoid; interneuron; opioid; optogenetics; parvalbumin; theta.
Figures
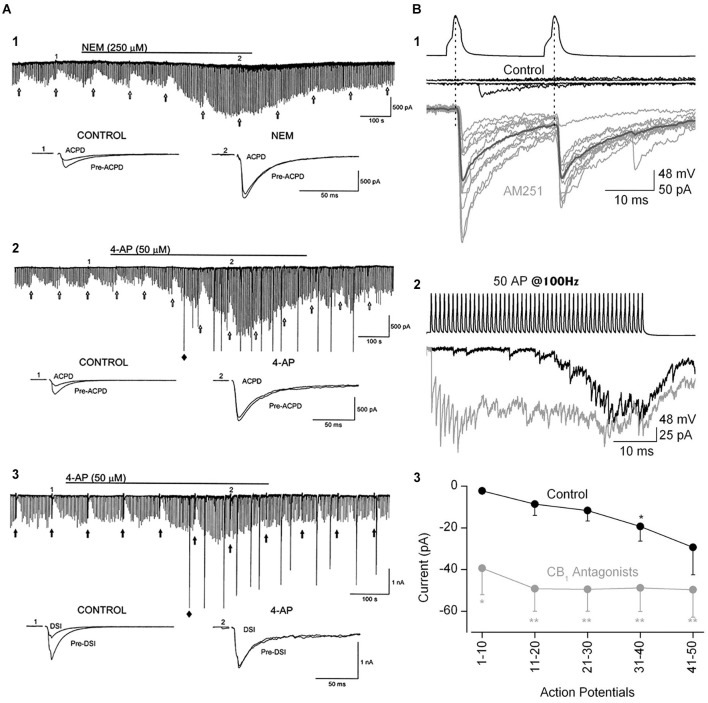
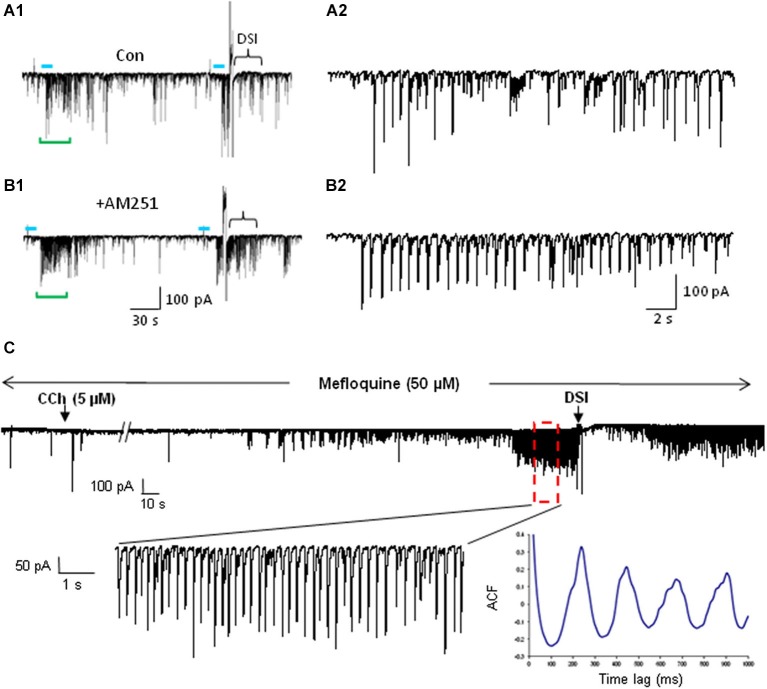
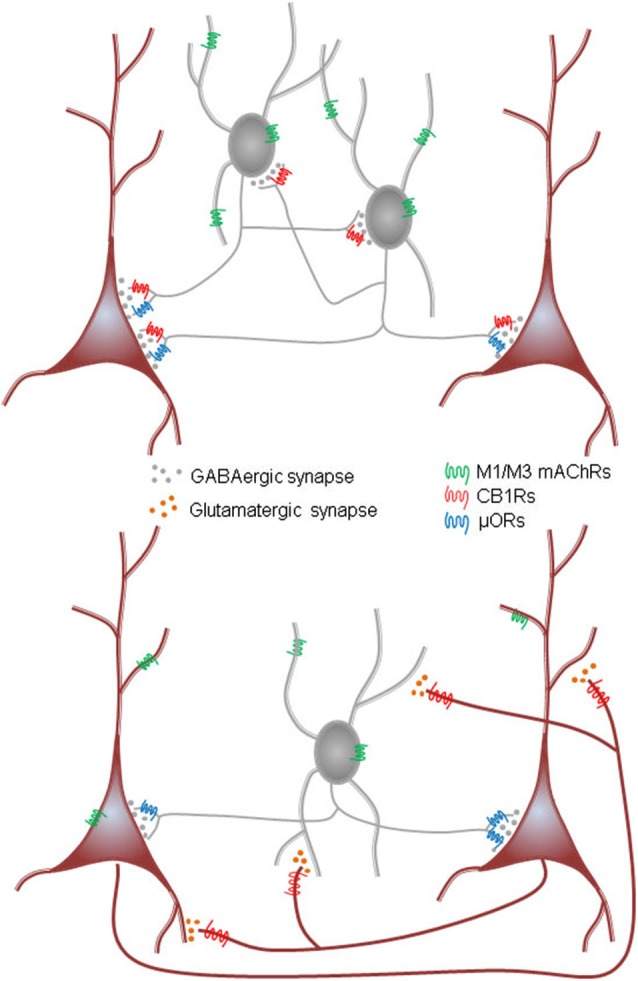
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources