

Characterization of the molecular mechanisms underlying increased ischemic damage in the aldehyde dehydrogenase 2 genetic polymorphism using a human induced pluripotent stem cell model system
- PMID: 25253673
- PMCID: PMC4215699
- DOI: 10.1126/scitranslmed.3009027
Characterization of the molecular mechanisms underlying increased ischemic damage in the aldehyde dehydrogenase 2 genetic polymorphism using a human induced pluripotent stem cell model system
Abstract
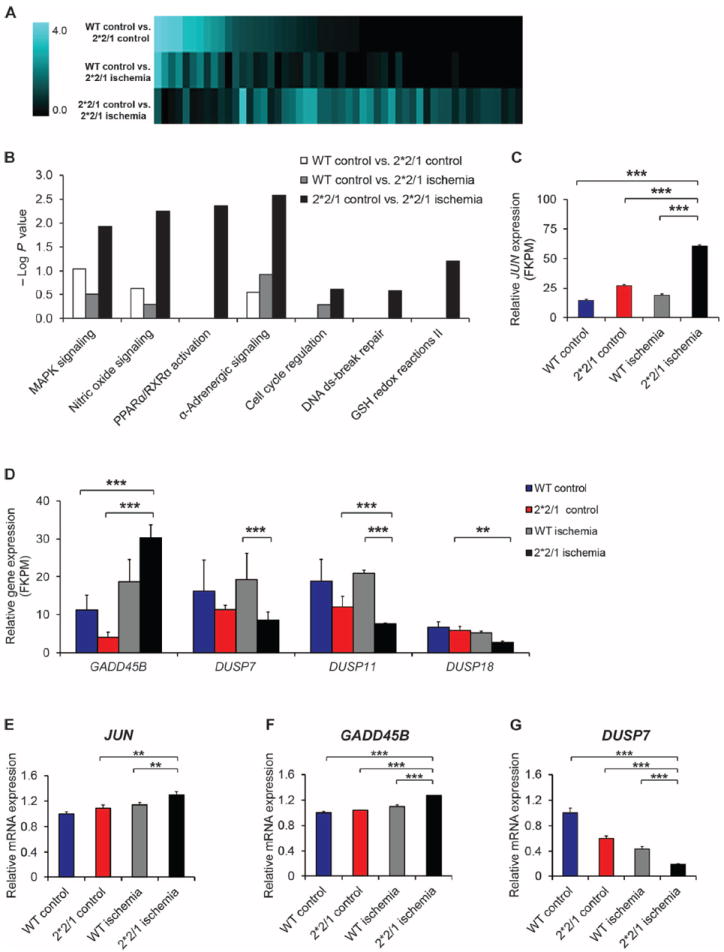
Nearly 8% of the human population carries an inactivating point mutation in the gene that encodes the cardioprotective enzyme aldehyde dehydrogenase 2 (ALDH2). This genetic polymorphism (ALDH2*2) is linked to more severe outcomes from ischemic heart damage and an increased risk of coronary artery disease (CAD), but the underlying molecular bases are unknown. We investigated the ALDH2*2 mechanisms in a human model system of induced pluripotent stem cell-derived cardiomyocytes (iPSC-CMs) generated from individuals carrying the most common heterozygous form of the ALDH2*2 genotype. We showed that the ALDH2*2 mutation gave rise to elevated amounts of reactive oxygen species and toxic aldehydes, thereby inducing cell cycle arrest and activation of apoptotic signaling pathways, especially during ischemic injury. We established that ALDH2 controls cell survival decisions by modulating oxidative stress levels and that this regulatory circuitry was dysfunctional in the loss-of-function ALDH2*2 genotype, causing up-regulation of apoptosis in cardiomyocytes after ischemic insult. These results reveal a new function for the metabolic enzyme ALDH2 in modulation of cell survival decisions. Insight into the molecular mechanisms that mediate ALDH2*2-related increased ischemic damage is important for the development of specific diagnostic methods and improved risk management of CAD and may lead to patient-specific cardiac therapies.
Copyright © 2014, American Association for the Advancement of Science.
Conflict of interest statement
Figures





References
-
- Guo YJ, Chen L, Bai YP, Li L, Sun J, Zhang GG, Yang TL, Xia J, Li YJ, Chen XP. The ALDH2 Glu504Lys polymorphism is associated with coronary artery disease in Han Chinese: Relation with endothelial ADMA levels. Atherosclerosis. 2010;211:545–550. - PubMed
-
- Takagi S, Iwai N, Yamauchi R, Kojima S, Yasuno S, Baba T, Terashima M, Tsutsumi Y, Suzuki S, Morii I, Hanai S, Ono K, Baba S, Tomoike H, Kawamura A, Miyazaki S, Nonogi H, Goto Y. Aldehyde dehydrogenase 2 gene is a risk factor for myocardial infarction in Japanese men. Hypertens Res. 2002;25:677–681. - PubMed
-
- Takeuchi F, Yokota M, Yamamoto K, Nakashima E, Katsuya T, Asano H, Isono M, Nabika T, Sugiyama T, Fujioka A, Awata N, Ohnaka K, Nakatochi M, Kitajima H, Rakugi H, Nakamura J, Ohkubo T, Imai Y, Shimamoto K, Yamori Y, Yamaguchi S, Kobayashi S, Takayanagi R, Ogihara T, Kato N. Genome-wide association study of coronary artery disease in the Japanese. Eur J Hum Genet. 2012;20:333–340. - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
