

The Alzheimer disease protective mutation A2T modulates kinetic and thermodynamic properties of amyloid-β (Aβ) aggregation
- PMID: 25253695
- PMCID: PMC4223304
- DOI: 10.1074/jbc.M114.599027
The Alzheimer disease protective mutation A2T modulates kinetic and thermodynamic properties of amyloid-β (Aβ) aggregation
Abstract
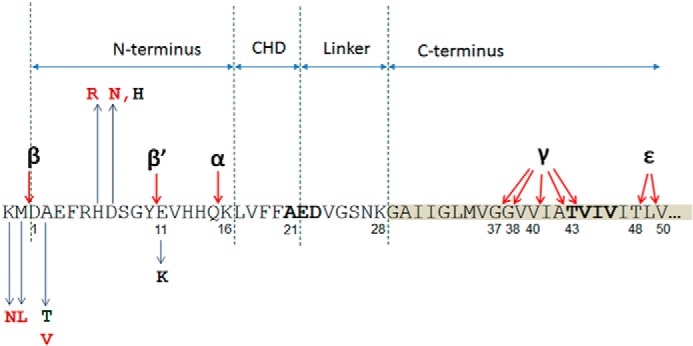
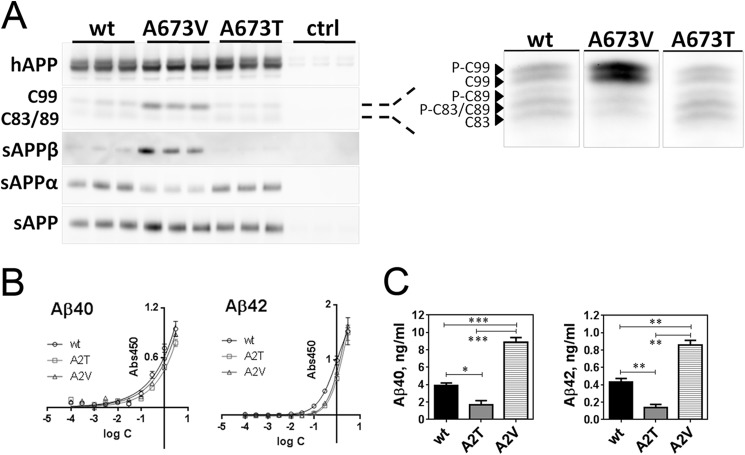
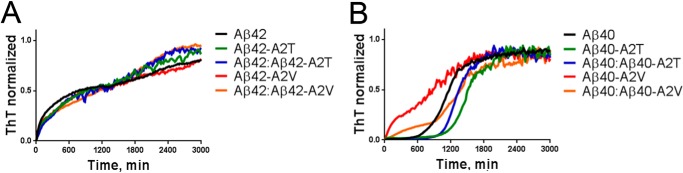
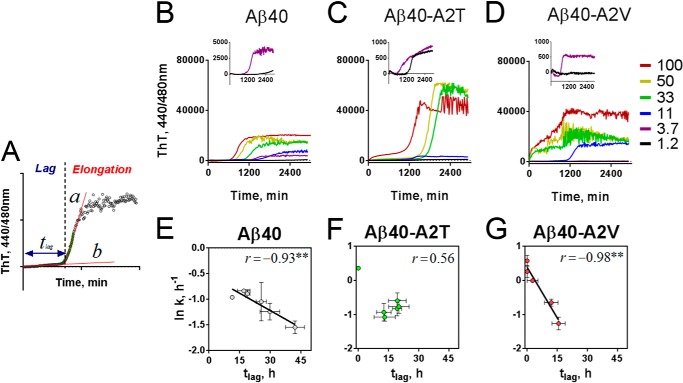
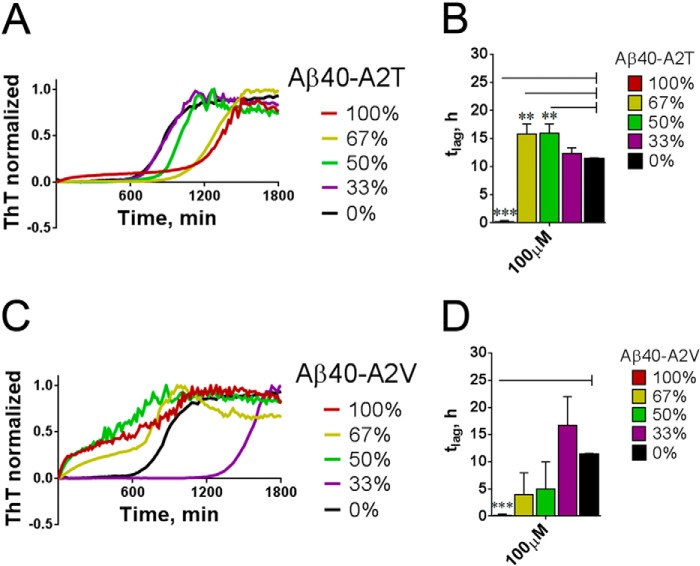
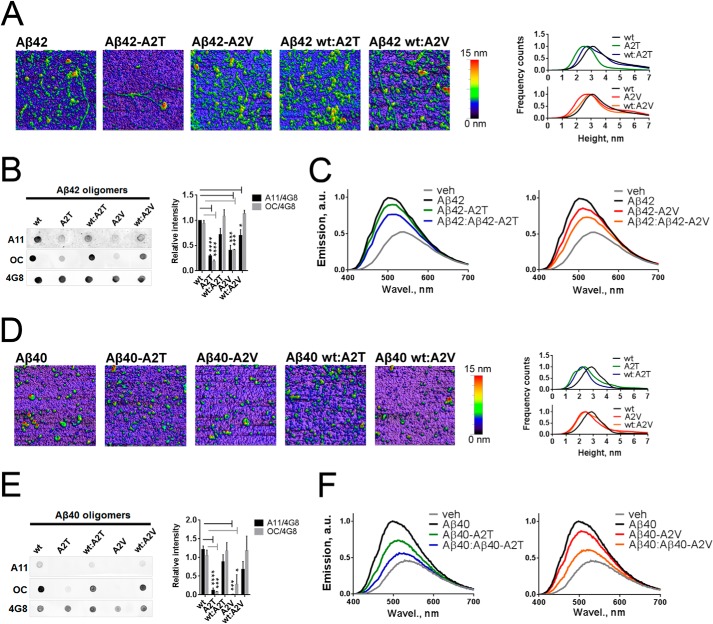
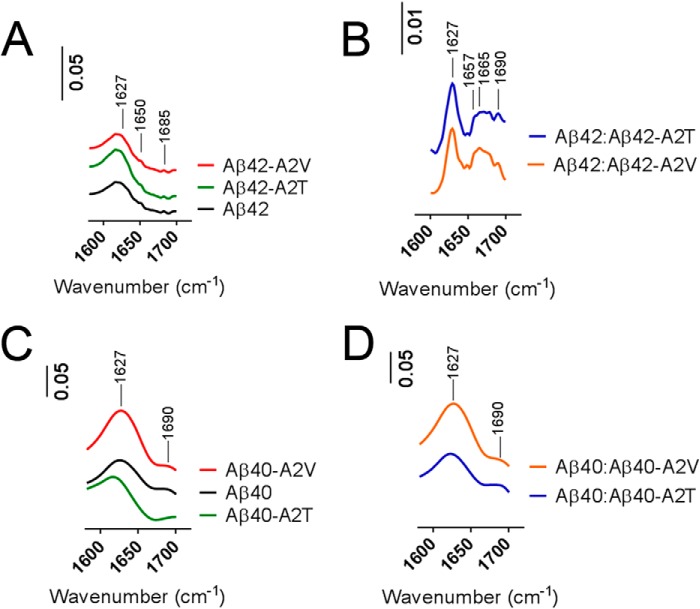
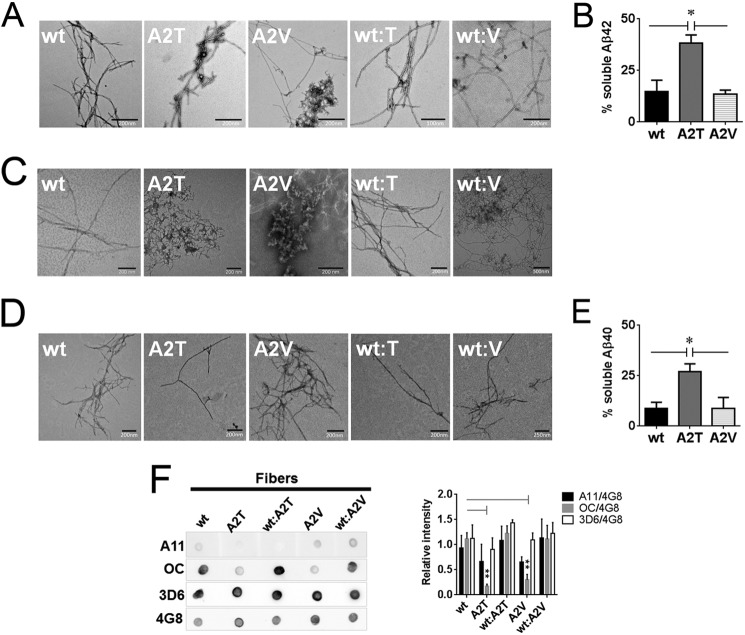
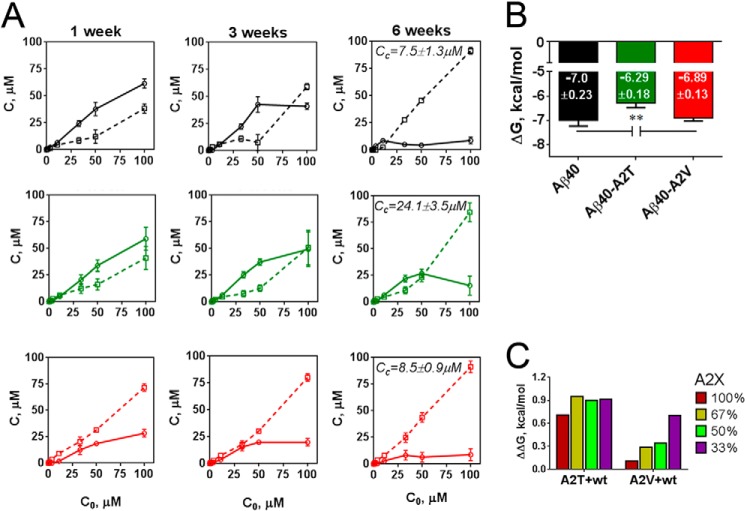
Missense mutations in alanine 673 of the amyloid precursor protein (APP), which corresponds to the second alanine of the amyloid β (Aβ) sequence, have dramatic impact on the risk for Alzheimer disease; A2V is causative, and A2T is protective. Assuming a crucial role of amyloid-Aβ in neurodegeneration, we hypothesized that both A2V and A2T mutations cause distinct changes in Aβ properties that may at least partially explain these completely different phenotypes. Using human APP-overexpressing primary neurons, we observed significantly decreased Aβ production in the A2T mutant along with an enhanced Aβ generation in the A2V mutant confirming earlier data from non-neuronal cell lines. More importantly, thioflavin T fluorescence assays revealed that the mutations, while having little effect on Aβ42 peptide aggregation, dramatically change the properties of the Aβ40 pool with A2V accelerating and A2T delaying aggregation of the Aβ peptides. In line with the kinetic data, Aβ A2T demonstrated an increase in the solubility at equilibrium, an effect that was also observed in all mixtures of the A2T mutant with the wild type Aβ40. We propose that in addition to the reduced β-secretase cleavage of APP, the impaired propensity to aggregate may be part of the protective effect conferred by A2T substitution. The interpretation of the protective effect of this mutation is thus much more complicated than proposed previously.
Keywords: Aggregation; Alzheimer Disease; Amyloid-β (Aβ); Kinetics; Thermodynamics.
© 2014 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
References
-
- Karran E., Mercken M., De Strooper B. (2011) The amyloid cascade hypothesis for Alzheimer's disease: an appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 10, 698–712 - PubMed
-
- Hardy J., Bogdanovic N., Winblad B., Portelius E., Andreasen N., Cedazo-Minguez A., Zetterberg H. (2014) Pathways to Alzheimer's disease. J. Int. Med. 275, 296–303 - PubMed
-
- Mullan M., Crawford F., Axelman K., Houlden H., Lilius L., Winblad B., Lannfelt L. (1992) A pathogenic mutation for probable Alzheimer's disease in the APP gene at the N terminus of β-amyloid. Nat. Genet. 1, 345–347 - PubMed
-
- Di Fede G., Catania M., Morbin M., Rossi G., Suardi S., Mazzoleni G., Merlin M., Giovagnoli A. R., Prioni S., Erbetta A., Falcone C., Gobbi M., Colombo L., Bastone A., Beeg M., Manzoni C., Francescucci B., Spagnoli A., Cantù L., Del Favero E., Levy E., Salmona M., Tagliavini F. (2009) A recessive mutation in the APP gene with dominant-negative effect on amyloidogenesis. Science 323, 1473–1477 - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
