

Folding simulations for proteins with diverse topologies are accessible in days with a physics-based force field and implicit solvent
- PMID: 25255057
- PMCID: PMC4195377
- DOI: 10.1021/ja5032776
Folding simulations for proteins with diverse topologies are accessible in days with a physics-based force field and implicit solvent
Abstract
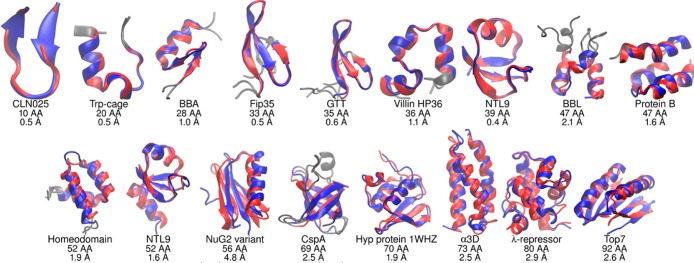
The millisecond time scale needed for molecular dynamics simulations to approach the quantitative study of protein folding is not yet routine. One approach to extend the simulation time scale is to perform long simulations on specialized and expensive supercomputers such as Anton. Ideally, however, folding simulations would be more economical while retaining reasonable accuracy, and provide feedback on structure, stability and function rapidly enough if partnered directly with experiment. Approaches to this problem typically involve varied compromises between accuracy, precision, and cost; the goal here is to address whether simple implicit solvent models have become sufficiently accurate for their weaknesses to be offset by their ability to rapidly provide much more precise conformational data as compared to explicit solvent. We demonstrate that our recently developed physics-based model performs well on this challenge, enabling accurate all-atom simulated folding for 16 of 17 proteins with a variety of sizes, secondary structure, and topologies. The simulations were carried out using the Amber software on inexpensive GPUs, providing ∼1 μs/day per GPU, and >2.5 ms data presented here. We also show that native conformations are preferred over misfolded structures for 14 of the 17 proteins. For the other 3, misfolded structures are thermodynamically preferred, suggesting opportunities for further improvement.
Figures
References
-
- RCSB Protein Data Bank; RCSB: Piscataway, NJ, 2014; Vol. 2014.
-
- Dill K. A.; MacCallum J. L. Science 2012, 338, 1042. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
